



INTRODUCCIÓN
El cáncer de paratiroides es una enfermedad poco frecuente1. El primer reporte fue hecho por De Quervain en 1909 (fig. 1), quien describió un paciente con una masa en el cuello que parecía paratiroides en la evaluación histopatológica2; posteriormente Amstrong, en 1938, describió la asociación de la hipercalcemia severa con este diagnóstico3. La etiología no se encuentra definida, aunque se ha asociado con ciertos síndromes endocrinos, y al parecer con irradiaciones previas en el cuello. El diagnóstico de esta entidad es difícil, no existen criterios definidos para establecer el diagnóstico previo al procedimiento quirúrgico y se han descrito ubicaciones muy raras de esta neoplasia4-6. El tratamiento es primordialmente quirúrgico, aunque se han hecho avances en la quimioterapia y radioterapia como terapias adyuvantes.

Fig. 1. Fritz de Quervain (1868-1940) cirujano suizo, gran autoridad en la enfermedad tiroidea, trabajó en Basel, Bern y Neuchâtel durante su carrera. Publicó numerosos artículos sobre enfermedad tiroidea, combinó en estos estudios conocimientos de epidemiología y técnica quirúrgica de la tiroidectomía. Su libro Special Surgical Diagnosis es un gran legado para la cirugía.
ESTRATEGIA DE BUSQUEDA DE LA LITERATURA
Se realizó una búsqueda en MedLine (1964-2006) usando los términos «carcinoma», «parathyroid», y «cancer»; la búsqueda inicial fue limitada al idioma inglés; se seleccionaron sólo artículos originales, omitiendo revisiones y otro tipo de literatura secundaria.
EPIDEMIOLOGÍA
El carcinoma de paratiroides es una neopla sia endocrina extremadamente rara1,4-7. En general el carcinoma del paratiroides se distribuye en forma similar por sexo8-11, a diferencia de la lesiones benignas de paratiroides que son más frecuentes en mujeres8,12; la edad de diagnóstico se encuentra entre los 15 y 81 años9, y son más frecuentes alrededor de la quinta década de la vida13; se han descrito pocos casos en personas jóvenes9. Por tanto, en algunos casos las consideraciones de género y edad son de poca ayuda en la evaluación individual del paciente. No existen diferencias en cuanto a la raza o grupos geográficos8,9,12.
ETIOLOGÍA
La etiología del carcinoma de paratiroides es hasta ahora incierta; ha sido asociada a formas esporádicas o familiares14, tales como la neoplasia endocrina múltiple tipo 1 (MEN-1), el síndrome de hiperparatiroidismo hereditario y el tumor de mandíbula (HPT-JT). Existe un riesgo incrementado en pacientes con hiperparatiroidismo familiar y antecedentes de irradiaciones15.
PATOGÉNESIS MOLECULAR
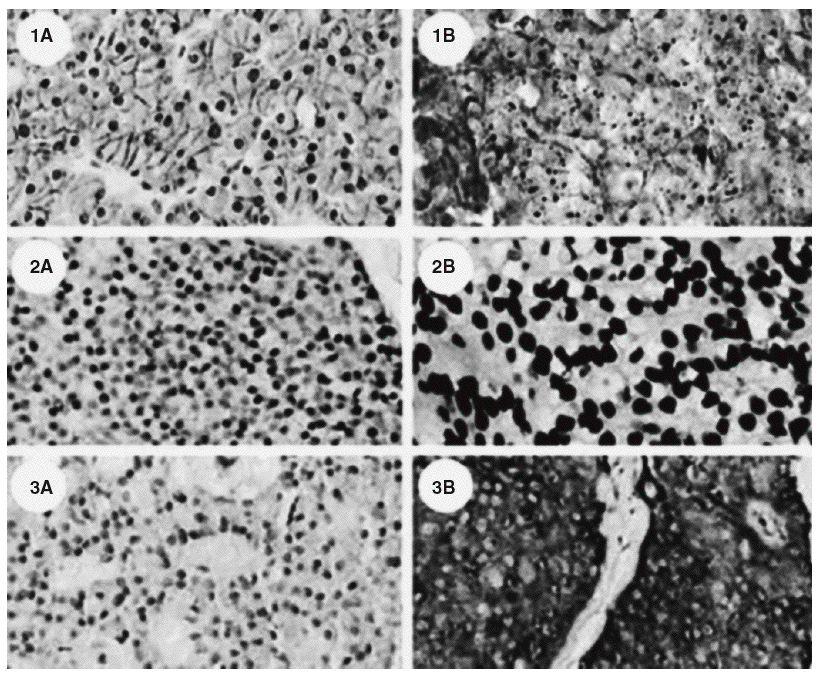
En la última década han aparecido nuevos conocimientos sobre las bases moleculares para el desarrollo de los tumores de las glándulas paratiroides. El primer gen en ser implicado fue el oncogén ciclina D1, cuyo papel es claro en el desarrollo de hiperplasias, adenomas y carcinomas de paratiroides14,16. Su expresión esta aumentada en el 40% de los adenomas y en un porcentaje mayor en los carcinomas17. El descubrimiento del gen del síndrome de la neoplasia endocrina múltiple tipo 1 (MEN-1) localizado en el cromosoma 11q1310 vino a confirmar una importante participación de este gen supresor tumoral en el desarrollo de tumores de paratiroides. Otro gen supresor tumoral envuelto en la carcinogénesis de la paratiroides es el gen del retinoblastoma o Rb 1, el cual se ha reportado en más casos de carcinoma que de adenoma14,18. A pesar de lo anterior el empleo de este marcador en la inmunohistoquímica para demostrar la presencia de cáncer no es seguro19. Recientemente se ha implicado también el gen HRPT2, previamente asociado con el síndrome de hiperparatiroidismo y tumores de la mandíbula y maxilares20,21. Este síndrome es conocido por presentarse en personas jóvenes con hiperparatiroidismo, tumores mandibulares y maxilares de carácter fibroso (no relacionados con el osteoide) y quistes renales22. Últimamente fue descrito el gen que codifica la proteína parafibromina, que sería un supresor tumoral23. Una descripción de las mutaciones del gen HRPT2 está asociada con malignidad en tumores de paratiroides; en muchos casos, las mutaciones son encontradas no sólo en forma somática, sino también sexual; esto abre nuevas perspectivas en la comprensión del carcinoma de paratiroides24,25. La descripción de que la parafibrina es una proteína controlada por la expresión de la ciclina D1 corrobora la hipótesis de la relación entre la mutación y el desarrollo de este tipo de neoplasias26; otra observación interesante es que las mutaciones de HRPT2 no están relacionadas con el desarrollo del hiperparatiroidismo familiar, sino con el hiperparatiroidismo, tumores de mandíbula y maxilares y carcinomas de paratiroides27. Esta entidad es extremadamente rara en pacientes con hiperparatiroidismo secundario o terciario28, lo que corrobora la hipótesis de que esta enfermedad se desarrolla en individuos predispuestos, lo que parece estar relacionado con mutaciones en HRPT2. Este hecho ayuda a dilucidar la definición anatomopatológica, ya que la inmunohistoquímica, empleando marcadores nucleares tales como E-cadherina, histona H1 y proteína precursora de amiloide bA4 podría definir el diagnóstico de cáncer29 (fig. 2), lo cual trae implicaciones en la conducta que se toma con los pacientes. En este sentido, cada caso de carcinoma de paratiroides deberá ser abordado como potencialmente representante de una condición hereditaria o familiar, y el estudio genético deberá ser importante y fundamental en estos casos.
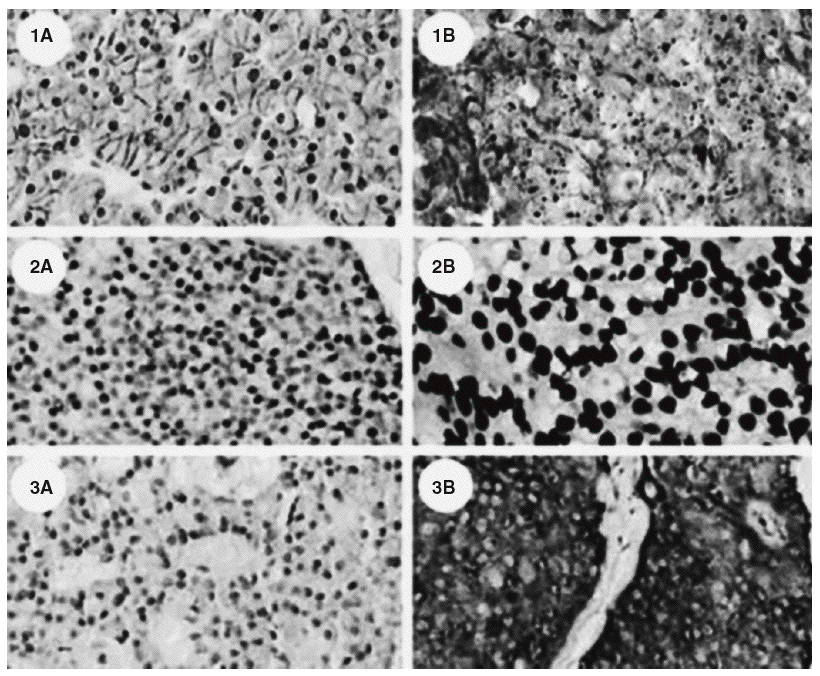
Fig. 2. Diferencia inmunohistoquímica entre (A) adenomas y (B) carcinomas paratiroideos; caracterizada por la sobreexpresión de E-cadherina (1), histona H1 (2), y proteína precursora de amiloidebA4 (3). Tomada de Haven CJ, Howell VM, Eilers PH, Dunne R, Takahashi M, van Puijenbroek M, et al. Gene expression of parathyroid tumors: molecular subclassification and identification of the potential malignant phenotype. Cancer Res. 2004;64:7405-11.
PRESENTACIÓN CLÍNICA
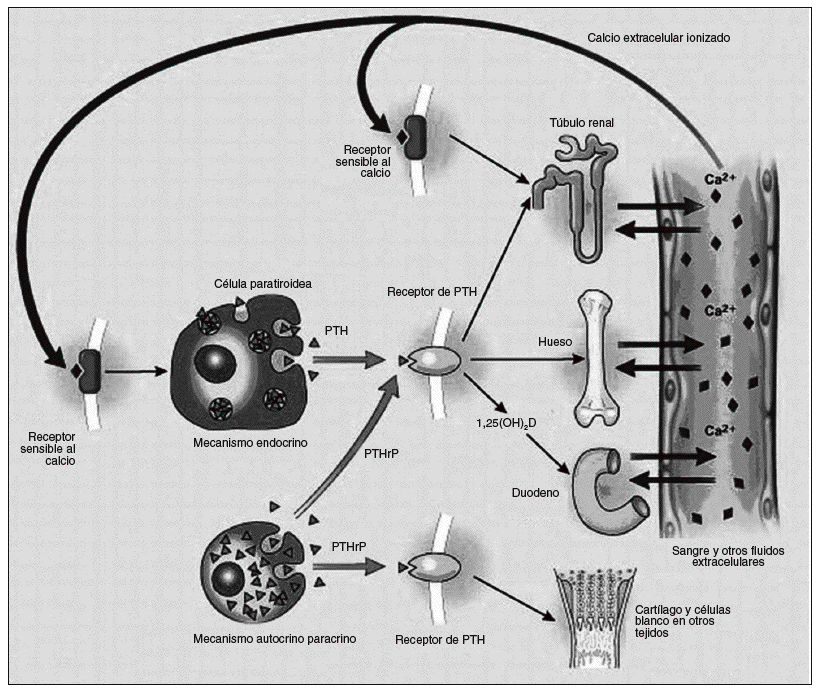
La mayoría de los carcinomas de paratiroides son funcionantes1,8, sin embargo existen algunos casos raros que no lo son30. Las manifestaciones clínicas se deben al hiperparatiroidismo primario e hipercalcemia más que a la infiltración a órganos vecinos11,31 (fig. 3). La mayoría de los pacientes están sintomáticos en el momento del diagnóstico (> 95%), al contrario de lo que sucede con las causas benignas de hiperparatiroidismo primario, en las cuales más del 80% se consideran asintomáticos1. Los órganos que con mayor frecuencia están seriamente comprometidos son el riñón y el hueso8. Según Wynne et al en una serie de casos1 la prevalencia de nefrolitiasis fue de 56%, y la de insuficiencia renal fue del 84%, a diferencia de los casos del hiperparatiroidismo (HPT) benigno en los cuales sólo del 4 al 18% manifiestan enfermedades renales9. Del 44 al 91% de los pacientes con carcinomas presentan manifestaciones óseas, tales como dolor, fracturas, osteítis fibrosa, resorción subperióstica y osteopenia difusa; contrario a esto menos del 5% de los pacientes con HPT benigno presentan alteraciones óseas y signos radiológicos específicos9,10. El 15% presentan pancreatitis, mientras que en el hiperparatiroidismo benigno ocurre en el 1,5%32.
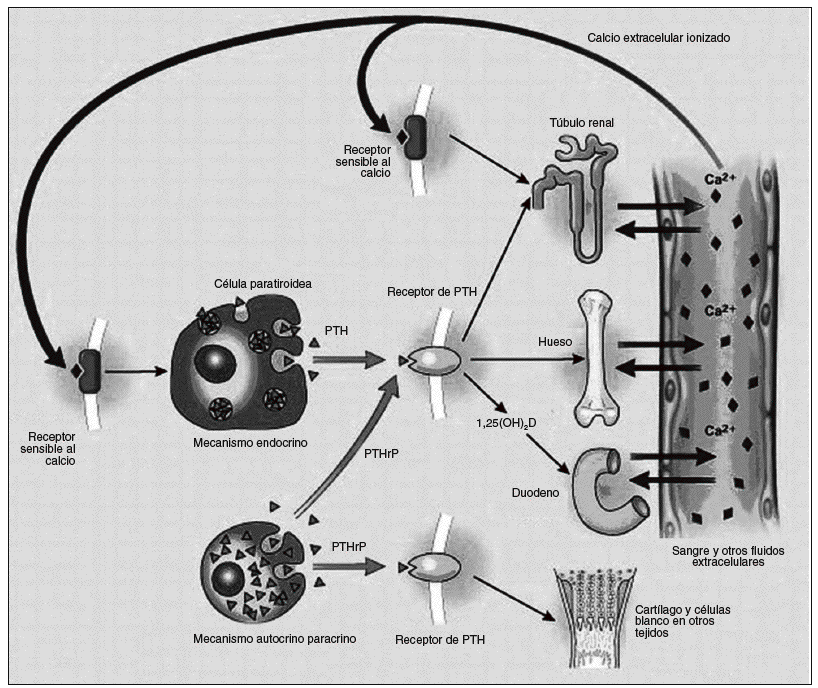
Fig. 3. El eje paratiroideo. La síntesis de hormona paratiroidea (PTH) y el péptido relacionado con la hormona paratiroidea (PTHrP) se muestran a la izquierda y sus sitios blanco de acción son mostrados a la derecha. Ellos actúan por medio del mismo receptor (que es llamado receptor PTH tipo 1). La retroalimentación negativa de la 1,25 dihidroxivitamina D no es mostrada. Un exceso o deficiencia de PTH puede traer las consecuencias nombradas en el texto.
El examen cuidadoso del cuello puede revelar la presencia de una masa firme entre el 30 y el 76% de los pacientes con carcinoma8. Este hallazgo es extremadamente raro en las otras causas de HPT primario32. En el 10% de los casos presentan disfonía por la compresión o invasión del nervio laríngeo recurrente31. Otros síntomas que están presentes en la mayoría de casos de carcinomas incluyen: fatiga, debilidad, polidipsia, poliuria, mialgias, artralgias, depresión, alteraciones del sueño, pobre memoria y manifestaciones gastrointestinales como anorexia, náuseas, vómitos, estreñimiento, dolor abdominal y úlcera péptica1. Aunque usualmente el crecimiento tumoral es lento, el curso clínico puede variar en cada paciente; esto depende de la agresividad del tumor y el grado de hipercalcemia8. Otros pacientes tienen poca o ninguna invasión local del tumor en el momento de presentación y experimentan una progresión más lenta, con escasa sintomatología9. Del 17 al 32% de los pacientes en el momento del diagnóstico presentan metástasis a nódulos linfáticos9.
ESTUDIOS DE LABORATORIO
Los niveles de calcio sérico están significativamente elevados en prácticamente todos los casos de carcinoma de paratiroides, con un valor promedio de entre 15 y 16 mg/dl ó 3-4 mg/dl por encima del límite superior normal, comparado con valores de 11 a 12 mg/dl de calcio sérico reportados en adenomas de paratiroides1. Igualmente, los niveles de hormona paratiroidea están muy elevados en comparación con las enfermedades benignas de la glándula; más del 70% de los pacientes tiene niveles 5 veces por encima del límite superior normal para la PTH8.
ESTUDIO HISTOPATOLOGICO
En el año de 1973 Schantz y Castleman definieron criterios histológicos para su diagnóstico; éstos son: patrón trabecular, figuras mitóticas, bandas fibrosas gruesas e invasión vascular y sanguínea33. Bodenson en 1993 redefinió los criterios histológicos para el diagnóstico, los cuales fueron presencia de crecimiento invasivo, macronucleolos y actividad mitótica34.
ESTADIFICACIÓN
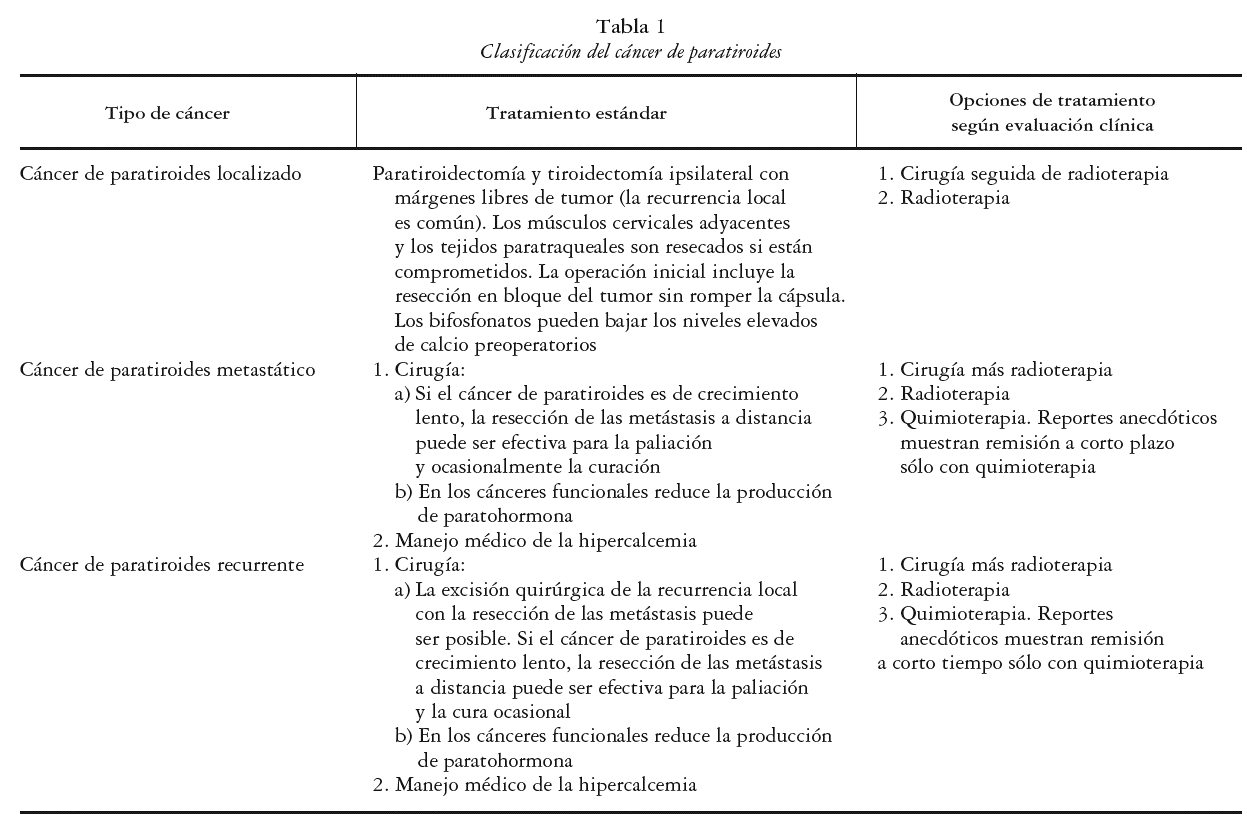
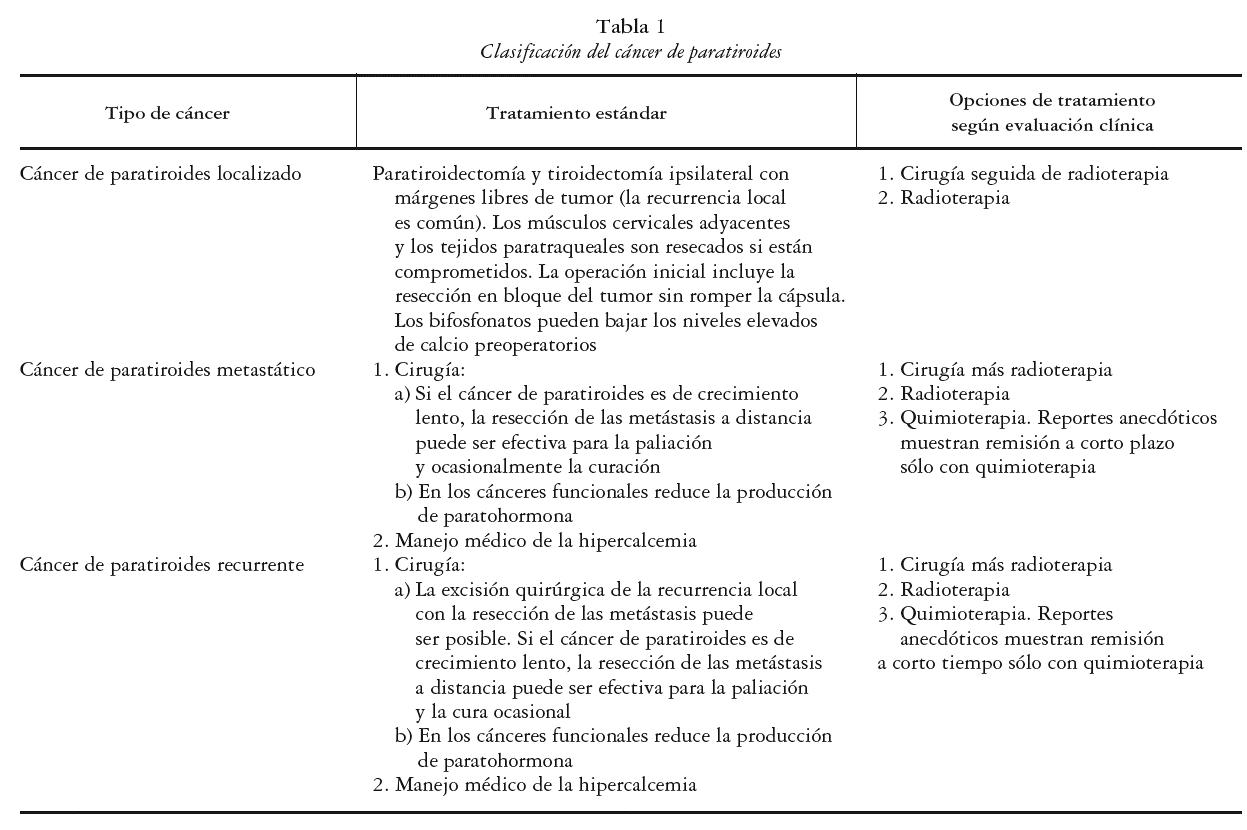
La estadificación del cáncer de paratiroides es compleja; la AJCC TNM (del inglés American Joint Committee in Cancer-Tumor Node Metastases), no es aceptada para el estadiaje de esta enfermedad. En la actualidad la enfermedad es clasificada en: localizada, metastásica o recurrente. Es localizada cuando el cáncer compromete a la glándula paratiroides con o sin invasión de tejidos adyacentes; metastásica si se presenta una invasión a los ganglios linfáticos regionales (la más frecuente), o si se encuentra en otros sitios más distantes como el pulmón, hueso, páncreas e hígado, y recurrente cuando reaparece con posterioridad a un control inicial de la enfermedad. Esto último se presenta en el 50% de los pacientes, y puede ocurrir hasta 34 años después del diagnóstico inicial. La clasificación se muestra en la tabla 1.
TRATAMIENTO
TRATAMIENTO MÉDICO
Debido a la presentación inusual del carcinoma de paratiroides, la eficacia de la quimioterapia y la radioterapia ha sido difícil de demostrar.
El uso de quimioterapia con dacarbazine (DTIC), sólo o en combinación con 5-fluorouracil (5-FU) y ciclosfosfamida ha dado pocos resultados. Al igual que el uso de otros fármacos, como la doxorrubicina y vincristina, ha dado poca o ninguna respuesta35.
La radioterapia como tratamiento primario no ha mostrado utilidad, aunque su uso en el cuello después de la cirugía puede ayudar a prevenir la recurrencia local y mejorar la sobrevida36.
Los pacientes con hipercalcemia no controlada, y quienes no recibieron tratamiento quirúrgico, deben recibir medicamentos que disminuyan los niveles de calcio sérico; dentro de las opciones se cuenta con los calcimiméticos, bifosfonatos, calcitonina y corticoides36. Los bifosfonatos disminuyen los niveles de calcio al actuar a nivel de la matriz ósea inhibiendo la resorción ósea mediada por osteoclastos. El uso de octreotide puede inhibir la secreción de PTH. En el 2004 la Food and Drug Administation (FDA) aprobó la cinacalcet (sensipar) para el tratamiento de la hipercalcemia asociada con carcinoma de paratiroides; éste es un agente calcimimético que actúa uniéndose a un sitio en los receptores de calcio extracelular, aumentando la afinidad del receptor por el calcio, disminuyendo la secreción de PTH, y posteriormente, descendiendo los niveles de calcemia36. Otros estudios con agentes calcimiméticos han demostrado que son capaces de disminuir el calcio sérico en pacientes con carcinoma de paratiroides metastásico en un período de dos años36.
La inmunoterapia también ha hecho su aporte; el uso de péptidos sintéticos de hormona paratiroidea humana o bovina ha demostrado que mediante una reacción mediada por células induce la producción de autoanticuerpos contra la PTH; este efecto normaliza los niveles de PTH, mejora la sintomatología y logra la remisión tumoral37.
TRATAMIENTO QUIRURGICO
Cuando se tiene un cuadro clínico que sugiera la presencia de un carcinoma de paratiroides, asociado a hipercalcemia y/o a una masa en el cuello, el único tratamiento que ha mostrado real beneficio es el manejo quirúrgico38. Provoca controversia el uso del BACAF para el diagnóstico, debido al riesgo de diseminación iatrogénica39. El manejo quirúrgico indicado es la resección en bloque de la lesión40, teniendo cuidado de preservar la cápsula del tumor, asociado a lobectomía de tiroides ipsilateral, escisión de tejido paratraqueal y los nódulos linfáticos (aunque el compromiso metastático a los nódulos linfáticos cervicales es menor al 20%). Se recomienda la escisión del grupo ganglionar linfático traqueo-esofágico, incluso la resección del nervio laríngeo recurrente se debe llevar a cabo si se observa compromiso de éste, evaluando las consecuencias de esta decisión40.
Se ha determinado que con la resección en bloque la supervivencia a 5 años es del 89%41, con una tasa de recurrencia del 8%; si se utiliza una paratiroidectomía simple la tasa de supervivencia disminuye a un 53% y la recurrencia se presenta en un 51% de los casos41. Se recomienda la toma de biopsia por congelación intraoperatoria para comprobar la presencia de tejido paratiroideo, no para evaluar las características neoplásicas42. Si el procedimiento quirúrgico se lleva a cabo y el diagnóstico se obtiene en el postoperatorio por histopatología, la reexploración del cuello está indicada con resección en bloque38.
Posterior al procedimiento quirúrgico se debe hacer seguimiento de los niveles de calcio y fósforo, debido a que una de las principales consecuencias de la resección del tumor es una hipocalcemia sintomática, denominada «síndrome del hueso hambriento», que debe ser manejada con suplementos de calcio y vitamina D; a su vez este síndrome es el mejor indicador de una resección completa de la masa y un procedimiento exitoso43,44.
El seguimiento de los pacientes a largo plazo se lleva a cabo controlando los niveles de calcio y de PTH43; los valores de calcio aumentados pueden deberse con más frecuencia a la presencia de recurrencia local y con menos frecuencia a las metástasis, las cuales se presentan en pulmón, hígado y hueso con mayor frecuencia30; cuando se sospeche recurrencia o metástasis deben realizarse pruebas de imagen como tomografía computarizada, resonancia magnética nuclear y gammagrafía con sestamibi; cuando éstas son identificadas, la resección quirúrgica constituye el manejo indicado, como medida de control de la hipercalcemia.
PRONÓSTICO
Los pacientes que son sometidos a cirugía de HPT primario y se les hace diagnóstico postquirúrgico de carcinoma de paratiroides tienen un peor pronóstico8.
El tiempo promedio entre la cirugía y la primera recurrencia es de 2 a 5 años en el 40-60% de los pacientes45. La localización más común de recurrencia se presenta en los tejidos del cuello y los nódulos linfáticos cervicales. En el 25% de los pacientes se produce metástasis a distancia, principalmente al pulmón, aunque también tiene lugar en el hígado y el hueso después de dos años de seguimiento46. En 1999 The National Cancer Data Base, en una serie de 286 casos, reportó una sobrevida a 5 años en el 85,5% de los mismos y una sobrevida a 10 años en el 49,1%47.
Finalmente se concluye que el cáncer de paratiroides debe tenerse en cuenta dentro del diagnóstico diferencial en casos de hiperparatiroidismo primario, en pacientes con masas palpables en cuello y/o hipercalcemia severa. Un diagnóstico temprano permite mejores desenlaces en este tipo de pacientes.