


Se trata de una paciente de 74 años de edad, con una historia de lumbociática izquierda diagnosticada en octubre de 1996 apreciándose en una resonancia magnética importantes degeneraciones de todos los discos lumbares con osteofitos posteriores, así como cambios degenerativos en varios espacios intervertebrales con rebordes osteocitarios y protrusión circunferencial de los discos que improntan el espacio subaracnoideo sin llegar a ocasionar una estenosis del canal tanto a nivel lumbar como dorsal. En los últimos meses presenta progresiva pérdida de fuerza con déficit motor para la marcha de manera que ha disminuido su actividad motora. También nota pérdida progresiva de la audición. Hay antecedentes de malformación ósea, posiblemente similares aunque no confirmadas directamente, en la familia.
En la exploración encontramos talla 147, peso de 55 kg, presión arterial (PA) de 120/80 mmHg; anormalidades faciales con aumento y abombamiento del frontal sobre el puente de la nariz, hipertolerismo ocular; estrechamiento de los pasajes nasales; sin signos de deterioro intelectivo asociado, y amaurosis bilateral.
También presenta nistagmus amaurótico, Romberg positivo con sensibilidad conservada, reflejos osteotendinosos exaltados, plantares indiferentes, clonus en ambas extremidades inferiores, sin otras alteraciones somáticas.
En la analítica practicada no se aprecian datos patológicos con una glucosa de 97, creatinina 0,7, colesterol 235. Serie hepática normal. Estudio metabólico con remodelado óseo igualmente normal (cociente Ca/Cr 0,0231, Hp/Cr 0,057, fosfatasa alcalina 99 UI/l y fosfatasa ácida tartrato-resistente 5,0 UI/l). El electromiograma muestra un patrón denervativo crónico de intensidad severa a nivel de los músculos tibial anterior y peroneo lateral así como de intensidad leve en gemelos.
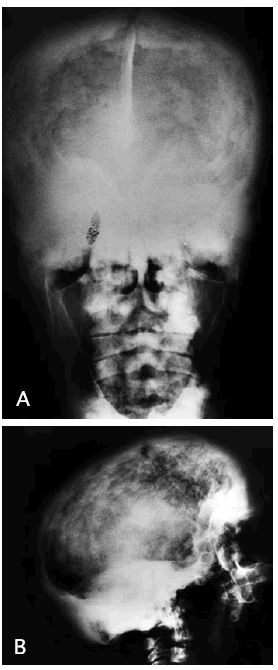
La radiografía de cráneo (fig. 1A y B) muestra importante engrosamiento de la calota craneal de manera generalizada a expensas fundamentalmente del diploe y la tabla interna. Esto condiciona una esclerosis progresiva de la base del cráneo con obliteración de los senos paranasales y pérdida de lámina dura dentaria. Con ello hay una disminución del tamaño de la cavidad craneal que puede producir compresión del parénquima encefálico, sobre todo a nivel de la fosa posterior, con cierta deformidad de las amígdalas cerebelosas a las que empuja hacia el tronco encefálico aunque no se observa hidrocefalia ni alteraciones que sugieran mielopatía compresiva. En la resonancia magnética cervical, se confirmaba la espondiloartrosis con posible mielopatía compresiva.
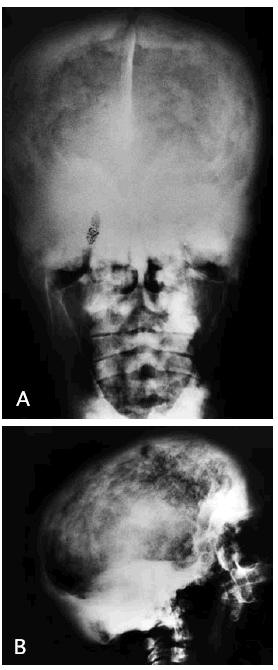
Fig. 1.
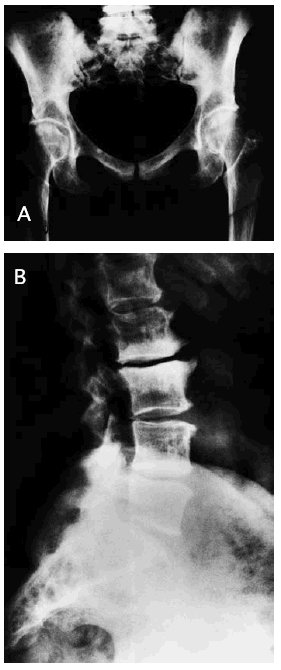
El resto del estudio radiológico óseo se apreciaba un aumento de la densidad ósea parcheada (fig. 2A y B) con deformidad tubular en la metáfisis de los huesos largos.
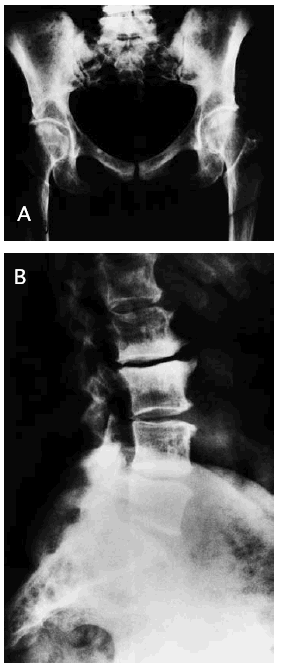
Fig. 2.
Las alteraciones faciales con pérdida de oído y defecto en la visión y posible presencia de algunas anormalidades en otros miembros de la familia sugieren el diagnóstico de displasia cráneo-metafisaria. Los datos básicos son la deformidad facial, la hiperosteosis craneal y un fallo al remodelado normal de los huesos tubulares. La forma recesiva de la enfermedad se acompaña de graves alteraciones faciales con hipertrofia de la masa ósea en la base de la nariz e hipertelorismo. También aparece maloclusión dental, parálisis facial, sordera y atrofia óptica y compresión de la masa cerebral. Aun cuando el pronóstico es bueno la participación compresiva neurológica puede afectar funciones motoras, como en nuestro caso, sensitivas y la calidad de vida.
El diagnóstico diferencial debe realizarse con la enfermedad de Pyle (displasia metafisaria), en que hay menos afectación del cráneo que del resto de los huesos tubulares, con la displasia cráneo-diafisaria y con la displasia diafisaria o enfermedad de Camurati-Engelmann.
NOTICIAS
LA FUNDACIÓN HISPANA DE OSTEOPOROSIS Y ENFERMEDADES METABÓLICAS ÓSEAS (FHOEMO)
Convoca el VI BECAS FHOEMO PARA JÓVENES INVESTIGADORES 2002 de acuerdo con las siguientes BASES
1. La Beca permitirá la realización de un período de aprendizaje y formación, sobre aspectos experimentales y/o clínicos en el campo de la Osteoporosis y Enfermedades Metabólicas Óseas, en un centro clínico especializado de ámbito nacional.
2. Las Becas estarán dotadas con 2.409,64 e.
3. Los candidatos serán posgraduados, menores de 40 años, interesados en el estudio de estas enfermedades.
4. La fecha límite de recepción será el 1 de junio de 2002.
5. La Beca será concedida por un Jurado que estará compuesto por cuatro miembros del Comité Científico de la FHOEMO, actuando como secretaria del Jurado, con voz pero sin voto, la Secretaria de la Fundación.
6. La propuesta consistirá en una concisa explicación sobre el proyecto, el Curriculum Vitae del solicitante y la aceptación del jefe de Servicio donde vaya a desarrollar su labor.
7. La documentación se remitirá a la Secretaría de la FHOEMO (Gil de Santivañes, 6, Bajo Int. D. 28001 Madrid. Tel/Fax: 91 578 35 10).
8. La decisión del Jurado será inapelable y las Becas podrán ser declaradas desiertas si, a juicio del jurado, se estimase que los proyectos presentados no reúnen los méritos suficientes.
9. Los aspirantes aceptarán las presentes bases por el mero hecho de concurrir a esta convocatoria.
BIBLIOGRAFIA RECOMENDADA
Gorlin R, Spranger J, Koszalka MF. Genetic craniotubular bone dysplasias and hyperostoses. A critical analysis. Birth Defects 1969;5:79-83.
McAlister WH, Hermann TE. Osteochondrodysplasias. Capítulo 88. En: Reswick D, editor. Diagnosis of bone and joint disorders. Philadelphia: Saunders; 1995.p.4208-10.