


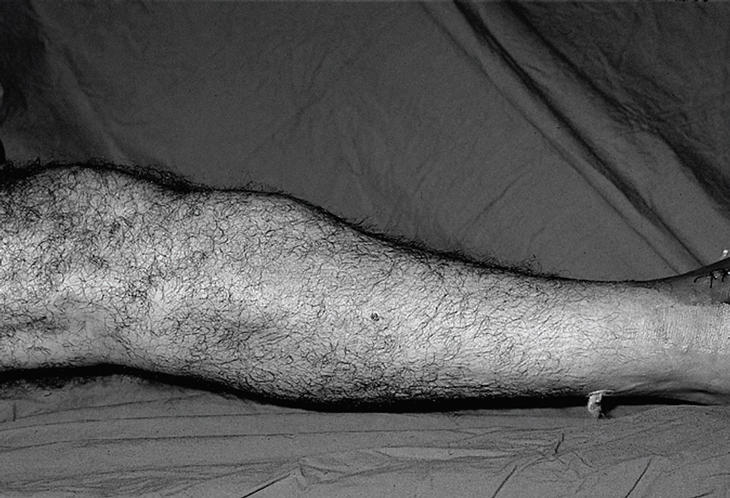
Se trata de un paciente varón de 45 años de origen magrebí, que consulta por tumoración y disminución de la movilidad con dolor en el pie derecho. Desde hace unos 15 años presenta tumoraciones localizadas en diáfisis femoral, tuberosidad tibial anterior y dorso del mediopié derecho, no dolorosas (fig. 1). Estas lesiones han aumentado de tamaño progresivamente, y le producen molestias en el pie y dificultad para caminar, así como una disminución de la movilidad, sobre todo a la flexión dorsal. Además, presenta inflamación vespertina en el tobillo derecho. Ha seguido tratamiento rehabilitador sin resultado. No refiere otros antecedentes de interés.
Figura 1. Tumoraciones localizadas en diáfisis femoral.
En la exploración destacan tumoraciones duras, no dolorosas a la palpación, localizadas en cara anteroexterna de diáfisis femoral y en tuberosidad tibial anterior de miembro inferior derecho. Tumoración dura en dorso del mediopié del mismo miembro. Limitación para la flexión dorsal del tobillo con un déficit de flexión de 10° y con una flexión plantar de 20°. Inversión y eversión prácticamente anuladas. Dolor a la palpación de articulación tibio-astragalina en cara anterior y de la articulación tibio-peronea anterior.
Los resultados del hemograma y de la bioquímica elemental fueron normales (incluidos calcio y fósforo) con excepción de un ligero aumento de la colesterolemia.
En las pruebas de imagen se observan los siguientes hallazgos:
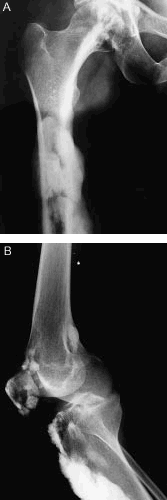
En la radiografía anteroposterior de fémur y cadera derecha (fig. 2 A) se observan imágenes de alta densidad de bordes lobulados en la región diafisometafisaria del fémur. En la radiografía lateral de rodilla derecha (fig. 2 B) se observan imágenes similares a las anteriormente descritas en la porción anterior de la tibia y periarticular.
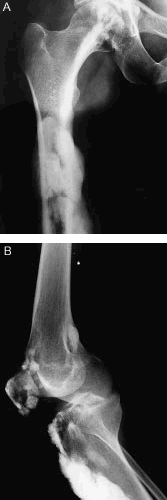
Figura 2. A. Radiografía anteroposterior de fémur y cadera derecha. B. Radiografía lateral de rodilla derecha.
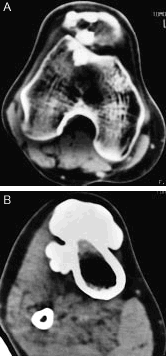
En la tomografía axial computarizada (TAC) de rodilla derecha (fig. 3 A) se observan calcificaciones periarticulares. En la TAC de la porción superior de la pierna derecha (fig. 3 B) destacan gruesas calcificaciones en la porción anterior de la tibia, de bordes lobulados.
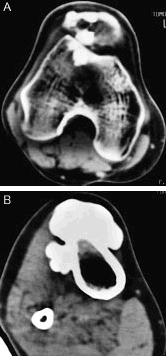
Figura 3. Tomografía axial computarizada de rodilla derecha (A) y porción superior de la pierna derecha (B).
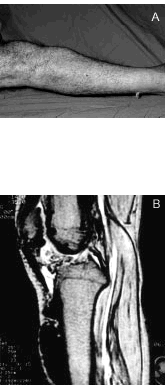
Se realizó resonancia magnética nuclear (RMN) de rodilla y pierna derechas en proyección sagital, en secuencias potenciadas en T1 (fig. 4 A) y T2 (fig. 4 B), donde las imágenes descritas son hipointensas en ambas secuencias.
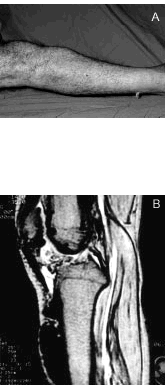
Figura 4. Resonancia magnética nuclear de rodilla y pierna derechas en secuencias potenciadas en T1 (A) y T2 (B).
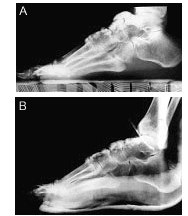
Por último, se realizaron radiografías laterales de pie derecho preoperatoria (fig. 5 A) y postoperatoria (fig. 5 B), donde se visualizan imágenes de similares características en el antepié, que fueron resecadas.
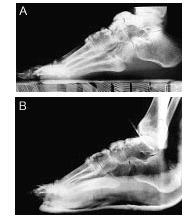
Figura 5. Radiografías laterales de pie derecho preoperatoria (A) y postoperatoria (B).
La intervención quirúrgica a la que fue sometido el paciente consistió en la resección de la neoformación localizada en el dorso del mediopié, con el fin de mejorar su sintomatología.
La pieza de resección fue enviada al servicio de Anatomía Patológica para su estudio. Consistía en tres fragmentos, el mayor de 3,5 x 2,5 x 0,8 cm, pétreos, irregularmente poligonales, y con parte de su superficie revestida por una fina lámina blanquecina nacarada, desflecada. A los cortes seriados, están constituidas por tejido óseo de aspecto compacto.
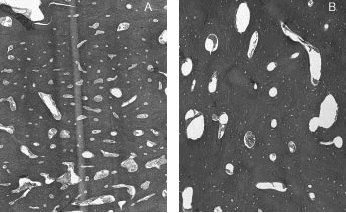
Histológicamente se aprecian amplias áreas de tejido óseo de aspecto cortical, con sistemas de Havers de distinto calibre, alguno de los mismos prácticamente obliterados (figs. 6 A y 6 B). En otras zonas se combina con hueso esponjoso.
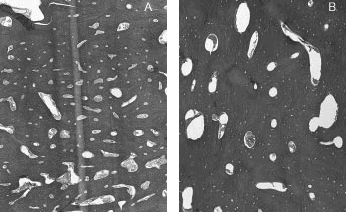
Fig. 6A y 6B. Estructura de los tejidos.
La observación bajo luz polarizada demuestra alternancia de hueso maduro, laminado, con áreas de hueso trenzado, con mínima irregularidad focal de líneas de cementación. Los espacios intertrabeculares están ocupados por tejido fibrovascular laxo.
La lesión, en su conjunto, se interpreta como una displasia ósea esclerosante, con participación de hueso de origen membranoso (perióstico) y encondral, aunque predomina la primera, confiriendo cierto aspecto semejante al apreciado en la enfermedad de Camurati-Engelmann. La distinción de ésta con la melorreostosis, e incluso un cuadro de solapamiento con otras osteodisplasias esclerosantes, se basa en la distribución característica radiológica de las lesiones óseas y de partes blandas.
DISCUSIÓN
La melorreostosis, también conocida como osteopatía hiperostótica u osteítis eburnizante monomiélica, fue descrita por primera vez por Leri y Joanny en el Bulletin Memoire de la Societé Médicale des Hopitaux de Paris en 19221, por lo que también se conoce como síndrome de Leri y Joanny.
Los casos más antiguos descritos en la literatura acerca de la melorreostosis aparecen en dos artículos. Uno de ellos data del 4.000-5.500 antes de J.C. y describe a una mujer de unos 25 o 35 años procedente del norte de Chile, originaria de la región del Morro que pertenece a la cultura del Chinchorro Chileno2. El otro caso descrito data del 500 después de J.C.3. En ambos casos, tanto la revisión como la comparación con la literatura médica, apoyan la hipótesis de que las lesiones que presentaban estos especímenes podrían corresponderse con los hallazgos descritos por Leri acerca de esta enfermedad.
Su nombre deriva del griego:μελφξ, melos (miembro),θεlv
En relación con la epidemiología de este proceso, no se han encontrado diferencias en su distribución por sexos. En cuanto a la edad de aparición, en el 50% de los casos la enfermedad se manifiesta antes de los 20 años, en ocasiones incluso debuta en los primeros días tras el nacimiento. De entre los casos cuyo diagnóstico fue más tardío, la mayoría padecían ya la enfermedad de forma silente años antes. Esto se explica por la lenta evolución de las lesiones que, si bien aparecen a edades tempranas, no dan clínica hasta aproximadamente el segundo decenio de la vida. Además, estas lesiones son poco típicas y difíciles de reconocer en el niño5,6. En cualquier caso, un inicio tardío de la enfermedad también es posible, como lo demuestra el primer caso de melorreostosis descrito por Leri, quien presentó a una mujer de 39 años con radiografías previas normales1.
En relación con la distribución de las lesiones, éstas suelen afectar a un solo miembro, por lo que se dice que tienen una distribución monomiélica, frecuentemente hemimiélica, siendo más común la participación del miembro inferior que la del miembro superior. Puede ser de carácter monostótico o poliostótico según afecte a uno o varios huesos de dicha extremidad7.
La esclerosis ósea aumenta con la edad, afectando a las diáfisis de huesos largos, huesos de la mano, pies y pelvis, pero en raras ocasiones afecta a las vértebras, costillas o epífisis óseas.
Las características distintivas de esta enfermedad son4:
1) Cambios escleróticos que están circunscritos a una extremidad.
2) Contorno del hueso afectado definitivamente distorsionado.
3) Dolor, que a menudo es intenso.
4) Limitación del movimiento en las articulaciones formadas por el hueso afectado.
En relación con la etiología de esta rara enfermedad existen varias hipótesis, pero actualmente se desconocen tanto las bases etiológicas como fisiopatológicas de la melorreostosis.
Campbell et al sugirieron en 19688 un posible origen congénito de inicio precoz durante el desarrollo embrionario y cronológicamente previo a la formación del esbozo de los miembros inferiores, que consistiría en una alteración de la hoja mesodérmica embrionaria.
En 1979 Murray y McRead9 observaron una estrecha relación entre la distribución de las lesiones de hiperostosis y los esclerotomos, zonas del esqueleto inervadas por un nervio sensitivo. Estos autores emitieron entonces la hipótesis de un posible origen infeccioso de etiología vírica similar al herpes zoster, dada la distribución lineal de las lesiones y la correlación topográfica con áreas de inervación sensitiva aislada. De los 30 casos estudiados por estos autores 19 presentaban lesiones de hiperostosis relacionadas con un único esclerotomo y las 11 restantes con múltiples esclerotomos, coincidiendo estos últimos casos con cuadros de melorreostosis clínicamente severos.
En 1980 McDougall et al10, dado que la melorreostosis es una enfermedad que afecta preferentemente al hueso, pero en la que se alteran los tejidos circundantes, deciden biopsiar el músculo que rodea las zonas de hiperostosis, encontrando en la microscopia electrónica cilindros compuestos de parejas membranosas organizadas en espirales y separadas entre sí por un sarcoplasma compuesto por glucógeno, mitocondrias y ocasionales cuerpos densos. Estructuras similares sólo habían sido descritas previamente en el músculo de dos pacientes. Uno de ellos padecía calambres musculares severos y había sido tratado con ciclofosfamida, clorambucil y prednisona por un linfoma linfocítico. El otro enfermo padecía una degeneración espinocerebelosa. Estos cilindros son similares ultraestructuralmente al retículo endoplásmico rugoso, afectando exclusivamente a fibras musculares tipo II, con un contenido importante de dicha organela. Se desconoce si este hallazgo es específico de la melorreostosis, pero cabría pensar en la posibilidad de una relación etiológica entre las anomalías óseas y musculares encontradas en los enfermos afectados por dicha enfermedad.
Burkus et al en 199711 y posteriormente Nervin et al en 19996 descubren dos casos de melorreostosis en pacientes afectados de osteopoiquilosis familiar, un trastorno autosómico dominante. La melorreostosis aislada o asociada a esta enfermedad aparece de forma esporádica. El hecho de que ambos trastornos se presenten de forma conjunta les hizo sospechar una posible relación genética entre ambas enfermedades. Estos autores sugieren que la melorreostosis de estos pacientes se debe a una mutación somática o a la pérdida del alelo homólogo normal en enfermos que padecen ostopoiquilosis, esto es, una segunda mutación en el mismo locus genético que origina la ostopoiquilosis.
Desde el punto de vista molecular, Kim et al en marzo de 200012 cultivaron fibroblastos procedentes de la piel de enfermos afectos de melorreostosis y de individuos sanos, e identificaron una diferente expresión génica a través de una hibridación tipo dot-blot. Estos autores encontraron que en los fibroblastos de los enfermos se expresaban en menor cantidad los genes que codifican por el factor 2 específico osteoblástico, las osteonectina, fibronectina y la bH3, cuyo gen inductor es el del TGF-b, siendo esta última proteína, cuya función es inhibir la formación de nódulos óseos de contenido osteoblástico, la más afectada. Estos hallazgos sugieren que una alteración en la expresión de estas moléculas de adhesión podría contribuir al desarrollo de hiperostosis y anormalidades concomitantes en los tejidos blandos, desempeñando el bH3 un papel preponderante en el seno de esta teoría etiológica.
Clínicamente, la melorreostosis se caracteriza por ser una enfermedad que cursa de manera asintomática durante un largo período dentro de su evolución, lo que condiciona el diagnóstico tan frecuentemente tardío de la misma y por tanto su detección hacia el segundo decenio de la vida.
En algunas ocasiones los pacientes pueden desarrollar cuadros de calambres musculares recurrentes y precoces en el seno del desarrollo de esta malformación ósea que siendo inespecíficos, son el único hallazgo sintomatológico previo a las manifestaciones clínicas típicas del cuadro, que sólo son evidentes en estadios avanzados del mismo.
En ocasiones dichos calambres musculares se asocian a una clara atrofia muscular de origen no filiado, pero probablemente de carácter compresivo y por tanto de etiología mecánica así como vascular.
El curso es generalmente crónico, con períodos de exacerbación y remisión. Las articulaciones y los tejidos blandos pueden verse afectados, apareciendo en ocasiones contracturas en flexión de las articulaciones del tobillo o del tendón de Aquiles, así como deformidades en genu varo o valgo de los miembros inferiores. También se ha descrito una anquilosis articular progresiva secundaria a la formación heterotópica de hueso y a las calcificaciones de los tejidos blandos, las cuales están en el origen de las alteraciones a nivel de la sensibilidad superficial y profunda de los miembros.
El cuadro florido de melorreostosis se acompaña de intenso dolor en la región afecta asociado a una impotencia funcional que imposibilita al individuo en sus actividades cotidianas. La melorreostosis dejada a su evolución puede condicionar graves deformidades, que no sólo están en el origen de la incapacidad funcional del paciente, sino que podrían acarrear alteraciones en el crecimiento del miembro, así como trastornos psicológicos secundarios a la aberración estética que producen.
Los hallazgos radiográficos de la melorreostosis son bastante característicos. La enfermedad suele aparecer en un solo miembro, dentro del cual puede afectar a uno o varios huesos.
Radiográficamente aparece una zona de hiperostosis con cortical engrosada, que se extiende a lo largo del hueso mostrando una imagen de «chorro de vela fluyendo». La hiperostosis del endostio es un rasgo asociado que puede obliterar total o parcialmente la cavidad medular13,14.
En los primeros momentos del proceso la articulación suele aparecer normal. Cuando avanza el curso de la enfermedad la hiperostosis puede llegar al margen de la articulación o incluso hacer protusión en la misma. En ocasiones aparecen islotes de tejido óseo en las epífisis de los huesos largos, o en los huesos del tarso. Es infrecuente la presentación de osificación en los tejidos blandos periarticulares, como rodilla, cadera, hombro, etc., y esta aparición suele darse en los pacientes más severamente afectados.
La TAC es una buena forma de demostrar el engrosamiento de la corteza. También es útil para diferenciar la osificación de tejidos blandos del tejido subyacente.
En la resonancia magnética (RM) podemos encontrar una baja señal en todas las secuencias (comportamiento consistente con el hueso cortical). Las masas paraarticulares se comportan de la misma manera13,14.
En la gammagrafía con Tc99 observamos un incremento de la captación del trazador en la zona afecta, que además preserva la cavidad medular15,16. La actividad puede cruzar la articulación para envolver huesos contiguos17. Esto puede ayudar a diferenciar la melorreostosis de otros procesos como osteopoiquilia y osteopatía estriada en los cuales no se produce aumento de la actividad15.
En cuanto a las asociaciones con otros procesos, en primer lugar, la melorreostosis ha sido relacionada con lesiones esclerodermatosas lineales, que consisten en bandas induradas de la piel, a menudo sobre áreas radiológicamente afectadas por melorreostosis. La biopsia de muestras de piel afectada revela áreas de fibrosis en el tejido subcutáneo o dermis18. Algunos autores se refieren a esta entidad como «esclerodermia lineal melorreostótica» y consideran que los cambios cutáneos son representativos del mismo desorden proliferativo que produce la hiperostosis de los huesos19. La hiperostosis cortical y la proliferación lineal del colágeno de la dermis profunda se han relacionado frecuentemente; lo que continúa sin esclarecerse es si esta entidad refleja una alteración metamérica común de ambas estructuras profundas, o si es la hiperostosis cortical la que provoca la proliferación del tejido subcutáneo y dermis profunda en las áreas vecinas20.
Los hallazgos sistémicos de esclerodermia no han sido asociados con la melorreostosis, sin embargo; hay descrito al menos un caso con melorreostosis y esclerodermia lineal que asociaba dismotilidad esófagica y fenómeno de Raynaud21.
La melorreostosis ha sido asociada también con algunos tumores: tres tumores lipomatosos han sido descritos en relación con la melorreostosis, dos de ellos localizados en la columna vertebral, uno intradural y otro intramedular, y el tercero en los tejidos blandos adyacentes al sacro22,23. También ha sido presentado un caso con desarrollo de fibromatosis «agresiva» (tumor desmoide) de los tejidos blandos adyacentes al área afectada por melorreostosis24.
Por otro lado, ha sido descrito un caso de osteosarcoma en relación con melorreostosis25. Se trataba de un paciente que desarrolló un osteosarcoma en el fémur derecho, habiéndole sido diagnosticado en esta localización una melorreostosis nueve años antes. En la discusión de este caso se propone que, como en otros procesos que se han asociado con osteosarcoma, la condición que favorezca el desarrollo de este tumor maligno sea el prolongado proceso de reparación por osteogénesis activa. Hay descrito otro caso de proceso maligno en relación con la melorreostosis, se trata de un histiocitoma fibroso maligno en relación con un área de melorreostosis del fémur26.
Ha sido publicado un caso de melorreostosis asociada a hipertensión renovascular e insuficiencia valvular aórtica27. Se ha propuesto que estas alteraciones fueran consecuencia del mismo trastorno, una displasia mesenquimal y vascular. En relación con esto también hay descritos casos que asocian melorreostosis y aneurismas arteriales28.
Por último, recientemente se ha descrito un caso de melorreostosis asociada a síndrome nefrótico de cambios mínimos y hemangiomas capilares29. Sus autores sugieren que la incidencia de anormalidades vasculares y de los tejidos blandos asociadas a melorreostosis permiten considerar que el origen de todas estas anormalidades puede estar en un defecto mesenquimal congénito producido durante el desarrollo de los sistemas vascular y musculoesquelético en la vida embrionaria.
El diagnóstico diferencial de la melorreostosis hay que hacerlo principalmente con las enfermedades asociadas con lesiones hiperostóticas30. Las más importantes a tener en cuenta van a ser: osteopetrosis, osteopoiquilia, osteopatía estriada, hiperostosis cortical infantil y los osteomas, que pertenecen al grupo de los tumores óseos benignos.
La osteopetrosis es una displasia ósea de etiología desconocida, que sigue un patrón hereditario autosómico dominante, en la que predomina una alteración de la osificación endocondral. El principal defecto es una disminución de la resorción de la esponjosa primaria, con aumento de la actividad de los osteoclastos, de manera que la resorción del cartílago endocondral está marcadamente disminuida, mientras que la formación de hueso continúa su ritmo normal. Esto se traduce en la radiología en una radioopacidad generalizada de la porción medular del hueso y una cortical característicamente ilesa, aunque en los huesos largos la esclerosis difusa puede dificultar la diferenciación entre cortical y medular31. Se pueden ver afectados todos los huesos, localizándose principalmente en las metáfisis. Es también frecuente en las vértebras, con la característica imagen en jersey de rugby, y en el anillo pélvico, con bandas en forma de anillos convexos asemejando una piel de cebra. A veces aparece una estriación longitudinal o transversal en fémur, tibia, peroné y metacarpianos. De esta manera las principales características que nos harían diferenciarla de la melorreostosis serían, en principio, que en esta entidad se trata de una alteración de la osificación endocondral mientras que la melorreostosis es una displasia ósea mixta que afecta tanto a la endocondral como a la intramembranosa, siendo más importante esta última. En la osteopetrosis hay un aumento de la actividad de los osteoclastos, mientras que en la melorreostosis se ve alterada la función de los osteoblastos. También es importante para diferenciarlas el hecho de que la cortical esté ilesa en la osteopetrosis, siendo esta zona la parte más afectada en la melorreostosis.
La osteopoiquilia es un trastorno autosómico dominante en el que se ve alterada la osificación endocondral igual que en el caso de la osteopetrosis, pero afectándose aquí la esponjosa secundaria, traduciéndose en un fallo en el remodelado óseo, con persistencia de trabéculas óseas maduras. Se asocia frecuentemente con lesiones en la piel del tipo de las dermatofibrosis, con fibromas en la espalda, brazos y piernas. La osteopoiquilia consiste en la aparición de múltiples islotes óseos agrupados en los extremos de los huesos, adyacentes a las metáfisis. Radiológicamente encontramos pequeñas opacidades simétricas y difusas en la zona yuxtaarticular de los huesos largos y en los huesos del tarso y del carpo. Es raro que aparezca en la columna, en el cráneo o en las costillas. Hay tres patrones característicos: ovalado o medular, lineal o estriado y mixto. Cuando aparece el patrón mixto se piensa que en realidad se trata de la coexistencia de osteopoiquilia, osteopatía estriada y melorreostosis.
La osteopatía estriada es también un trastorno autosómico dominante que forma parte de las displasias de la osificación endocondral, e igual que la osteopoiquilia, con afectación de la esponjosa secundaria. Afecta a todos los huesos excepto el cráneo y la clavícula, y tiene predilección por los huesos largos (fémur distal, tibia proximal, húmero proximal). El diagnóstico generalmente es debido a un hallazgo casual en la radiología, siendo lo más característico unas estriaciones lineales densas, que representan el fallo de remodelado del hueso maduro, con persistencia de éste. La imagen característica consiste en líneas paralelas a lo largo del hueso y ocasionalmente cruzando las epífisis.
En algún caso las imágenes radiológicas pueden hacernos confundir estas dos entidades con la melorreostosis32,33. Por ello lo que tendremos en cuenta cuando veamos lesiones en las epífisis y en tarso y carpo, que son las zonas de osificación endocondral, es el hecho de que aparezcan lesiones también en las metáfisis y la simetría. Todo esto es propio de la osteopoiquilia y la osteopatía estriada. Para el diagnóstico diferencial definitivo la gammagrafía con Tc99 es muy útil, ya que en la melorreostosis hay captación del radioisótopo, mientras que no la hay en las otras dos entidades.
Los osteomas34 pertenecen al grupo de los tumores óseos benignos. Son lesiones anormalmente densas que se han formado a partir del periostio. Las zonas más frecuentes son el cráneo y la cara, aunque también pueden aparecer en la pelvis y en los huesos tubulares de las extremidades. La edad más frecuente de aparición es entre la cuarta y la quinta décadas de la vida. Son asintomáticos y radiológicamente se caracterizan por aparecer como focos radiodensos, únicos o múltiples, que se extienden desde la superficie del hueso de origen. Tienen contorno liso y son homogéneos.
Por último podríamos tener en cuenta la hiperostosis cortical infantil, que es otro trastorno autosómico dominante, que aparece en los primeros meses de la vida y se acompaña de tumefacción de partes blandas. Consiste en una hiperostosis cortical, con depósitos de hueso en el tejido blando inmediatamente contiguo a la cortical original. La localización más frecuente es en mandíbula, clavícula, costillas, ilíacos, cráneo y tubulares (el cúbito el más frecuente). Es característica una mejoría radiológica progresiva e incluso la desaparición de la lesión en semanas o meses.
En estas dos últimas patologías, teniendo en cuenta la edad de aparición, la localización y el aspecto de las lesiones, es fácil hacer el diagnóstico diferencial. Es importante en el caso de la hiperostosis cortical infantil su curso progresivo hacia la autorresolución. Esto nunca ocurre en la melorreostosis, salvo raras excepciones35.
Con estos datos podemos llegar a la conclusión de que los puntos importantes para hacer el diagnóstico diferencial en la melorreostosis serían: la falta de evidencia de tratarse de un desorden genético; la edad de aparición de las manifestaciones; el curso progresivo crónico; la afectación de articulaciones y partes blandas vecinas, y anatomopatológicamente, afectación de osificación intramembranosa, con aumento de actividad de los osteoblastos, así como la característica imagen fluente en la radiología.
La melorreostosis es generalmente un proceso asintomático. Cuando aparecen los síntomas lo hacen en forma de dolor e impotencia funcional en las articulaciones que se ven afectadas por el crecimiento progresivo de la lesión. Cuando el único síntoma es el dolor es suficiente aplicar medidas conservadoras como antiinflamatorios no esteroideos (AINE) u otros analgésicos menores. Existe algún caso tratado con nifedipino, con éxito, basándose en la teoría de una posible alteración vascular como patogénesis de la enfermedad36.
Otros muchos métodos, tanto conservadores como quirúrgicos, han sido utilizados para el tratamiento de la melorreostosis, tales como manipulaciones, férulas de escayola, cirugía para liberación de partes blandas, capsulotomías, osteotomías y artroscopia para eliminar fragmentos óseos intraarticulares37.
Ha sido descrito algún caso de dismetría de miembros en niños, debido a contracturas en flexión, en los que se ha utilizado con éxito métodos de distracción, como es el aparato de Ilizarov38,39.