




La osteomalacia es consecuencia de una alteración de la mineralización de la matriz ósea, que conduce al cúmulo de osteoide no mineralizado y disminuye la resistencia del hueso. Cuando el trastorno de la mineralización se produce en niños se afectan las placas epifisarias de crecimiento y aparece el raquitismo1.
ETIOLOGÍA
Para que se produzca la mineralización del osteoide se necesitan dos condiciones principales: por un lado, que haya calcio y fósforo en concentración suficiente; por otro, que no existan inhibidores de la mineralización1-3. La vitamina D es necesaria para mantener las concentraciones de calcio y fósforo; de hecho, la 1,25(OH)2D o calcitriol es un importante estimulador de la absorción intestinal de estos elementos. Se debate si además los metabolitos de la vitamina D facilitan la mineralización por una acción directa sobre las células óseas. En todo caso in vitro el calcitriol favorece la diferenciación de los osteoblastos y la síntesis de fosfatasa alcalina, y esta enzima sí es imprescindible para una mineralización normal de la matriz, pues hidroliza el pirofosfato, que es un inhibidor fisiológico de la mineralización.
Desde un punto de vista práctico los procesos causantes de osteomalacia y raquitismo se pueden dividir en tres grupos
(tabla 1):

1. Los que cursan con alteraciones de la vitamina D.
2. Las hipofosfatemias de otro origen (no relacionadas con la vitamina D), hereditarias o adquiridas.
3. Los que suponen una acumulación de inhibidores de la mineralización.
DEFICIENCIA DE VITAMINA D (OSTEOMALACIA NUTRICIONAL)
En nuestro medio, la mayor parte de los casos de osteomalacia se deben a una deficiencia de vitamina D4. La síntesis cutánea inducida por las radiaciones ultravioleta es la fuente principal de vitamina D. Por tanto, la deficiencia de vitamina D suele aparecer en individuos con pobre exposición solar.
Otros factores que pueden conducir a la deficiencia de vitamina D son los tratamientos con antiepilépticos (que aceleran el catabolismo de la vitamina D al inducir la formación de metabolitos polares inactivos en el hígado); las hepatopatías graves (en las que disminuye la capacidad de síntesis de 25[OH]D), y los síndromes de malabsorción intestinal (en los que no sólo disminuye la absorción de la vitamina D de la dieta, sino que se altera la circulación entero-hepática de metabolitos de la vitamina D que son normalmente excretados en la bilis y rebasorbidos posteriormente).
RAQUITISMOS HEREDITARIOS
Estos trastornos se presentan generalmente en la infancia, en forma de raquitismo, pero en ocasiones pueden aparecer como una osteomalacia en la edad adulta. Los «raquitismos dependientes de la vitamina D» se deben a alteraciones en el metabolismo de la vitamina D o sus receptores5. Los «raquitismos resistentes a la vitamina D» se deben a hipofosfatemia secundaria a pérdidas renales de fosfato.
Raquitismo dependiente de la vitamina D tipo I (RVDD-I)
Se debe a una ausencia de 1-alfa-hidroxilasa renal, la enzima que convierte la 25(OH)D en 1,25(OH)2D. Se transmite con herencia autosómica recesiva.
Raquitismo dependiente de la vitamina D tipo II (RVDD-II)
Es una alteración genética del receptor de la vitamina D, necesario para la acción de la 1,25(OH)2D o calcitriol. Por tanto, también se conoce con el término de «raquitismo hereditario resistente al calcitriol». La herencia es autosómica recesiva. Las manifestaciones suelen aparecer en los dos primeros años de vida, aunque se han descrito casos esporádicos leves de aparición más tardía. Más de la mitad de los pacientes presentan alopecia. El tratamiento requiere dosis muy elevadas de calcio y calcitriol.
Raquitismos resistentes a la vitamina D
Se deben a la pérdida de fosfato por el riñón6.
1. Raquitismo hipofosfatémico ligado al cromosoma X. Es el más común de los «raquitismos resistentes a la vitamina D» (VDRR), suponiendo el 80% de los casos. La prevalencia está en torno a 5/100.000. Se debe a una mutación en el gen PHEX (phosphate regulating gene with homologies to endopeptidases on the X chromosome). Este gen, situado en el cromosoma X, codifica la síntesis de una metaloproteasa cuyo sustrato fisiológico no está plenamente identificado. Se expresa en el hueso y otros tejidos. La patogenia es compleja. En primer lugar, la deficiencia de PHEX parece provocar una alteración directa de la mineralización del hueso. En segundo término el PHEX sería responsable en condiciones normales de degradar un factor estimulador de la fosfaturia («fosfatonina»). Al faltar PHEX aumentarían los niveles de dicho factor, lo que se traduciría en un aumento de la fosfaturia, y en consecuencia, hipofosfatemia. En tercer lugar, dicho factor inhibiría además la síntesis de 1,25(OH)2D. Ese factor fosfatúrico (y eventualmente depresor de la síntesis de 1,25(OH)2D no ha sido identificado con certeza, pero varios estudios recientes sugieren que puede tratarse del FGF-23 (factor de crecimiento fibroblástico 23).
Se transmite ligado al cromosoma X, con carácter dominante. En general, las manifestaciones aparecen en los dos primeros años de vida, pero la expresividad clínica es variable y hay casos menos graves que se manifiestan como osteomalacia en la edad adulta.
2. Raquitismo hipofosfatémico autosómico. Parece deberse a mutaciones del gen que codifica el FGF-23, situado en el cromosoma 12, resultando un FGF-23 más resistente a la degradación por proteasas. Se transmite con carácter autosómico dominante, pero la penetrancia es incompleta y la expresión clínica variable. Algunos casos se manifiestan en la infancia, con características similares al raquitismo ligado al cromosoma X. Otros se manifiestan como osteomalacia en la edad adulta.
3. Raquitismo hipofosfatémico con hipercalciuria. Es muy raro, sólo se han descrito unas pocas familias. El trastorno genético responsable no ha sido bien aclarado, pero presumiblemente está relacionado con defectos en las proteínas implicadas en la reabsorción tubular renal del fósforo. La expresividad clínica es muy variable. A diferencia de otros raquitismos hipofosfatémicos cursa con aumento de la calciuria y de los niveles séricos de 1,25(OH)2D.
OSTEOMALACIA ONCOGÉNICA
Es un trastorno poco frecuente (unos 150 casos descritos en la literatura), asociado generalmente a tumores poco agresivos de origen mesenquimal. Las características clínicas y bioquímicas son similares a las de los raquitismos hipofosfatémicos hereditarios. Se supone que se debe a la producción excesiva de FGF-23 por las células tumorales7.
CLÍNICA
El hueso osteomalácico tiene menor resistencia y está predispuesto a las fracturas. Además, a menudo estos pacientes presentan dolores osteomusculares, aun sin fracturas evidentes.
Los síntomas miopáticos, sobre todo la debilidad muscular proximal, son frecuentes. Se deben a la hipofosfatemia y quizás a la pérdida del efecto directo de los metabolitos de la vitamina D sobre las células musculares4. Cuando existe hipocalcemia pueden aparecer manifestaciones tetánicas. A veces aparecen sinovitis o cuadros similares a la distrofia simpática refleja. La hipofostemia puede asociarse también a calcificaciones ligamentosas y sacroileítis, que pueden simular una espondilitis anquilosante8.
Los síntomas osteomalácicos, a veces acompañados de adelgazamiento, pueden ser la primera manifestación de un síndrome de malabsorción oculto4,9,10. Esa combinación de dolores óseos múltiples y pérdida de peso puede hacer sospechar equivocadamente la existencia de un tumor con metástasis óseas.
En los niños raquíticos se dan además deformidades óseas (genu valgo o varo, deformidades craneales y costales, etc.), alteraciones dentarias y retraso de crecimiento.
DIAGNÓSTICO
En la osteomalacia por deficiencia de vitamina D, que es la causa más frecuente de la misma, suelen existir niveles séricos de calcio bajos o normales-bajos, hipocalciuria, hipofosfatemia y aumento de fosfatasa alcalina y de la parathormona (PTH). Los niveles de 25(OH)D, que son los que mejor reflejan la dotación de vitamina D, están también bajos, pero los de 1,25(OH)2D son variables y a menudo normales. En nuestra experiencia4 la fosfatasa alcalina y la PTH intacta son los parámetros más sensibles para el diagnóstico, estando elevados en más del 90% de los pacientes (tabla 2).

La radiología suele mostrar hallazgos inespecíficos como osteopenia o pérdida de altura de los cuerpos vertebrales. Infrecuentes, pero más características de osteomalacia son las pseudofracturas o líneas de Looser-Milkman. En los niños raquíticos son típicas las alteraciones de las metáfisis, que aparecen ensanchadas e irregulares.
En las gammagrafías se observan focos hipercaptadores múltiples, que no deben confundirse con las metástasis.
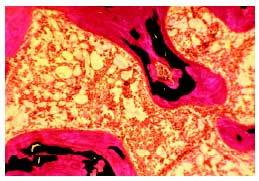
El diagnóstico definitivo de osteomalacia requiere la realización de una biopsia ósea, previa administración de tetraciclinas, que se fijan en los lugares que están siendo mineralizados. Se observa un aumento de las superficies osteoides y del grosor de las vetas de osteoide (fig. 1). En consecuencia, el volumen total de osteoide es muy superior al normal. El estudio histomorfométrico pone de manifiesto también un retraso de la mineralización (superior a 100 días). Pueden existir signos de aumento de la actividad osteoclástica, reflejo del hiperparatiroidismo secundario acompañante.
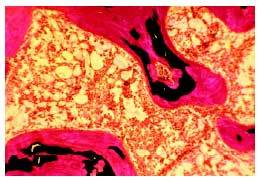
Fig. 1. Biopsia ósea de una paciente con osteomalacia secundaria a malabsorción intestinal. Se observa un marcado aumento del volumen de osteoide (teñido en color rojo), quedando pocas áreas de hueso adecuadamente mineralizado (teñido en negro).
En ausencia de biopsia ósea se puede establecer un diagnóstico de presunción de osteomalacia por deficiencia de vitamina D ante un cuadro clínico compatible acompañado por los siguientes datos:
1. Hipocalcemia, hipofosfatemia, aumento de fosfatasa alcalina o pseudofracturas.
2. Aumento de PTH o disminución de 25(OH)D.
3. Ausencia de insuficiencia renal.
4. Resolución tras tratamiento con vitamina D.
TRATAMIENTO
Si es posible se efectuará un tratamiento causal (tratamiento dietético de la malabsorción, etc.). Se debe asegurar una ingesta de calcio adecuada, de 1-2 g/día.
Las necesidades diarias de vitamina D están en torno a 400-800 U/día. En caso de osteomalacia por deficiencia de vitamina D se deben administrar cantidades mucho mayores, de unas 30.000-200.000 unidades semanales, en una o varias dosis, durante varios meses.
Los metabolitos hidroxilados de la vitamina D se absorben más fácilmente que la propia vitamina, por lo que pueden ser preferibles en presencia de malabsorción o colestasis. El calcidiol (25[OH]D) se utiliza en dosis de 50-150 microgramos/día; el calcitriol (1,25[OH]2D), en dosis de 0,5-1 microgramos/día.
Sin embargo, la respuesta individual es difícilmente predecible, por lo que se deben controlar periódicamente los niveles de calcio sérico y urinario, fosfato y fosfatasa alcalina, con el fin de comprobar la mejoría progresiva de las alteraciones previas y evitar la aparición de hipercalcemia o hipercalciuria.
Las osteomalacias por hipofosfatemia se tratan con suplementos de fosfato y calcitriol.