


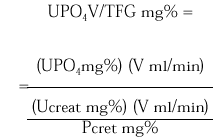

INTRODUCCION
En los últimos años se ha producido un notable progreso en nuestra comprensión de los desórdenes hereditarios de la pérdida urinaria de fosfato. El conocimiento ganado con el estudio de estos desórdenes ha permitido también tener una mayor comprensión del mantenimiento normal de la homeostasis del fosfato. En esta revisión primero examinaremos los mecanismos normales de transporte de fosfato por el túbulo renal, su regulación dietética y hormonal, describiremos los trastornos hereditarios de la pérdida urinaria de fosfato y finalmente haremos una breve descripción de su forma de diagnóstico.
MANEJO RENAL DEL FÓSFORO
La ingesta usual de fósforo está en el rango de 25 a 60 mmol/día (775-1.860 mg P/día). Entre sujetos normales que comen dietas que proveen 40 mmol/día (1.240 mg/día) la absorción intestinal neta es de alrededor del 60% (25 mmol/día o 775 mg/día) pudiendo llegar hasta el 80%. A pesar de que el hueso es remodelado diariamente y que el fósforo muscular es reciclado desde y hacia el líquido extracelular (LEC) en el curso del metabolismo, no hay ganancia ni pérdida neta de fósforo en estos procesos. Por lo tanto, para mantener un estado de equilibrio en que la ingesta se equilibre con la pérdida, el riñón deberá excretar 25 mmol de PO4 por día o 775 mg. Los niveles normales de fósforo sérico están en el rango de 0,9 a 1,5 mmol/l (2,8 a 4,6 mg%) siendo la concentración promedio 1,2 mmol o 3,7 mg%. Casi todo el fósforo del plasma es ultrafiltrable excepto un 10% que se encuentra formando complejos. Para un sujeto adulto cuya filtración glomerular es de 180 1/día, se filtran 6.500 mg de PO4 por día; si se excretan 775 mg/día, se puede ver que el 88% del PO4 es reabsorbido y sólo el 12% es excretado.
El grueso del fósforo filtrado es reabsorbido en el túbulo proximal, siendo aproximadamente el 60% de la carga filtrada reclamada en su porción convoluta y 15 a 20% en su porción recta. Sólo una pequeña y variable porción de la carga filtrada (< 10%) es reabsorbida en las porciones más distales del nefrón.
MECANISMOS CELULARES DE TRANSPORTE TRANSEPITELIAL DE FOSFORO EN EL TUBULO PROXIMAL: SISTEMAS DE COTRANSPORTE Na/P
El transporte transepitelial de fósforo es esencialmente unidireccional e involucra la entrada por la membrana apical con ribete en cepillo, su pasaje a través de la célula, y su salida por la membrana basolateral. La captación del PO4 a nivel de la membrana apical es el paso limitante de todo el proceso de reabsorción y es el sitio más importante de regulación1. La entrada de fosfato al túbulo proximal es mediada por transportadores de fosfato sodio dependientes localizados en la membrana apical, que a su vez dependen del gradiente de sodio creado por la Na/K ATPasa que se encuentra en la membrana basolateral para movilizar el proceso de transporte. El cotransporte Na/P es muy sensible a los cambios de pH, incrementándose de 10 a 20 veces cuando pH se eleva de 6 a 8,5. Esto refleja no sólo el transporte preferencial de la forma bivalente de fosfato, sino también la acción de los protones sobre el cotransporte. Por lo menos dos sistemas cinéticamente diferentes de cotransporte Na/PO4 han sido identificados en la membrana apical del túbulo proximal: uno de alta capacidad y baja afinidad, que se encuentra sólo en la porción convoluta y que es responsable del grueso del transporte de fosfato proximal, y otro sistema de baja capacidad y alta afinidad, que se encuentra tanto en la porción convoluta como recta y que es responsable de la reclamación residual de fosfato.
Recientemente se han identificado los cADN que codifican los cotransportadores Na/P específicos para el riñón de varias especies2. Basados en las secuencias de nucleótidos, los cADN codifican dos clases de cotransportadores Na/P que tienen sólo 20% de homología y han sido designados como NPT1 y NPT2 (símbolos aprobados por el Human Genome Mapping Workshop). El transportador NPT1 está compuesto de aproximadamente 465 aminoácidos que se despliegan 7a 9 veces en la membrana y está codificado en el cromosoma humano 6p22; asimismo, el transportador NPT2 está compuesto por aproximadamente 635 aminoácidos que se despliegan 8 veces en la membrana y está codificado en el cromosoma humano 5q353. Los estudios de localización renal han encontrado que el NPT1 está uniformemente expresado en todos los segmentos del túbulo proximal, la expresión del NPT2 es mayor en el segmento S14. Tanto el NPT1 como el NPT2 median el cotransporte Na/P de alta afinidad.
Recientemente se ha obtenido evidencia de la expresión de dos nuevos transportadores de PO4 en el riñón. Ambos son receptores virales de superficie: el G1vr-1 (gibbon ape lukemia virus) y el Ram-1 (murine amphotropic virus), que median un transporte de PO4 sodio dependiente electrogénico5. Poco se conoce acerca de la translocación del fosfato a través de la célula. Así los iones de PO4 que ingresaron a la célula se equilibran rápidamente con los pools intracelulares de fosfatos orgánicos e inorgánicos. También existen pocos datos acerca de la salida de PO4 por la membrana basolateral. Este último parece ser un proceso pasivo que depende del gradiente eléctrico a través de la membrana y ocurre por vía de un mecanismo de intercambio aniónico1.
REGULACION DE LA REABSORCION RENAL DE FOSFATO
REGULACION POR EL FOSFORO DE LA DIETA
La ingesta dietética de PO4 es un regulador muy importante de reabsorción renal de fosfato. La deprivación dietética de fosfato provoca un incremento en el máximo tubular para el fosfato que se atribuye a un incremento en el cotransporte Na/P a nivel de la membrana apical, y este incremento es independiente de factores extrarrenales. La señal para la respuesta adaptativa a la deprivación de fosfato no se conoce. Se ha sugerido que la caída en la concentración de fosfato en la célula renal juega un papel en mediar la respuesta de incremento del transporte. Se ha observado un incremento en la velocidad máxima de transporte (Vmax) tanto del sistema de cotransporte Na/P de alta capacidad baja afinidad como del de baja capacidad alta afinidad. La fase aguda de la respuesta adaptativa a la deprivación de fosfato (2 horas) se ha asociado a un incremento en la proteína NPT2 pero no del ARNm del NPT26. El incremento en el cotransporte Na/P y en la proteína NPT2 en la membrana se revierte rápidamente por una dieta alta en fosfato. Todos estos datos sugieren que la vía endocítica/exocítica está comprometida en mediar la fase aguda de la respuesta a la deprivación de fosfato. En contraste, en la deprivación crónica se ha observado un incremento en la abundancia renal tanto de la proteína NPT2 como de su ARNm6. Los estudios de deprivación crónica efectuados de fósforo en células de riñón de opossum han demostrado que el incremento del ARNm del NPT2 se debe a un incremento de la estabilidad de ARNm más que a un incremento de la transcripción génica7. Por otro lado, la expresión génica del NPT1 no se incrementa marcadamente con la restricción de fósforo, mientras que los transportadores Glvr-1 y el Ram son regulados por el fosfato extracelular tanto a nivel del ARNm como a nivel funcional.
REGULACION HORMONAL
Los cotransportes Na/P de la membrana apical del túbulo proximal son el blanco de regulación por una variedad de hormonas peptídicas, esteroideas y factores de crecimiento.
De las hormonas peptídicas la paratohormona (PTH) es la más importante de las hormonas reguladoras de la reabsorción de fosfato. La PTH actúa directamente sobre el túbulo proximal inhibiendo el transporte Na/P por mecanismos dependientes tanto del AMPc/protein kinasa A como de la fosfoinositoles/protein kinasa C8. La acción de la PTH es mediada por receptores tanto apicales como basolaterales. La acción de la PTH sobre el cotransporte Na/P es interrumpida por agentes que interfieren con la vía endocítica y se ha obtenido evidencia directa por inmunohistoquímica de internalización de la proteína NPT2 de la superficie apical de la célula en respuesta a la PTH9.
De las otras hormonas que intervienen en la regulación de la reabsorción del fosfato, la hormona de crecimiento, la insulina, el IGF I, las hormonas tiroideas y la 1,25(OH)2D3 estimulan la reabsorción, mientras que la PTHrP, la calcitonina, el AMP, el epidermal growth factor (EGF), la tasa de filtración glomerular (TFG) alfa y los glucocorticoides inhiben la reclamación de fosfato. Mientras que las hormonas tiroideas incrementan la abundancia del ARNm para el NPT210, tanto la dexametasona11 como el EGF12 disminuyen la abundancia del ARNm, por lo que es probable que el efecto tanto de las hormonas tiroideas como los glucocorticoides se ejerza a través de la regulación de la transcripción genética que producen estas hormonas al unirse a sus receptores específicos intracelulares.
Estudios recientes han descubierto dos nuevos sistemas hormonales en la regulación del fosfato. Una de ellas es la stanniocalcina, hormona antihipercalcemiante derivada del corpúsculo de Stannius de los peces, que se ha demostrado que estimula la reabsorción de fosfato en el túbulo proximal del lenguado13. La stanniocalcina se ha identificado en el túbulo contorneado distal y colector del riñón humano y se ha demostrado que reduce la excreción de fosfato cuando se infunde en la rata14. Estos hallazgos sugieren que la stanniocalcina puede contribuir al mantenimiento de la homeostasis del fosfato en los mamíferos al igual que en los peces. La otra hormona es un factor fosfatúrico aislado de tumores productores de osteomalacia. Cuando estos tumores son trasplantados a ratones atímicos, éstos presentan hipofosfatemia y pérdida urinaria de fosfato15. El medio de cultivo donde se desarrollan células de estos tumores inhibe el transporte de fosfato en células de riñón de opossum. Esta hormona o factor fosfatúrico ha sido denominado genéricamente fosfatonina y ha sido parcialmente purificada de un cultivo celular de un hemangioma esclerosante causante de osteomalacia tumoral16.
TRASTORNOS HEREDITARIOS DE LA PÉRDIDA URINARIA DE FOSFORO
Se han descrito tres hipofosfatemias hereditarias. Las tres tienen como anormalidad subyacente una disminución de la reabsorción renal de fosfato y cada una de ellas se asocia a raquitismo u osteomalacia. Estos tres desórdenes difieren en su modo en que se heredan, en la severidad de la enfermedad ósea, en el metabolismo de la vitamina D y en la respuesta a la terapéutica.
RAQUITISMO HIPOFOSFATÉMICO LIGADO AL X (XLH)
El XLH es un desorden hereditario dominante ligado al X con poco o ningún efecto de dosis génica17 y es la forma más frecuente de los trastornos hereditarios de pérdida de fosfato con una prevalencia de 1:20.000. Los pacientes presentan generalmente baja estatura, raquitismo con la resultante deformidad en las extremidades inferiores, dolor óseo, y enteropatía (calcificación de tendones, ligamentos y cápsulas articulares)18. Los individuos más afectados pueden tener anormalidades craneanas y estenosis del canal raquídeo. Un signo clínico frecuentemente pasado por alto es la apariencia de los dientes: mientras que el raquitismo hipocalcémico presenta hipoplasia del esmalte, el XLH tiene defectos de la dentina, que si bien no son aparentes al examen pueden causar abscesos dentarios. A pesar de la hipofosfatemia la debilidad no es una queja frecuente de estos pacientes. El elemento más importante de esta enfermedad es la pérdida renal de fosfato caracterizada por una reducción en el máximo tubular para el fosfato en relación a la tasa de filtración glomerular (TmP/GFR). Otras características de laboratorio incluyen un incremento en la fosfatasa alcalina total y ósea, normocalcemia, niveles normales de hormona paratiroidea y niveles de calcitriol inapropiadamente normales para el nivel de fosfato plasmático18.
Los estudios histológicos de biopsias óseas de cresta ilíaca muestran además de la osteomalacia una lesión periosteocítica hipomineralizada característica del XLH y que nunca desaparece completamente a pesar de que la mineralización activa restaure las superficies endostales19.
Existe un modelo murino de XLH, el ratón Hyp. Los estudios cinéticos en este ratón han demostrado que estos animales tienen un defecto en el sistema de cotransporte Na/P de alta afinidad y baja capacidad, existiendo un 50% de reducción tanto del ARNm como de la proteína del Npt2, homólogo murino del NPT2 humano20. Además de la pérdida de fosfato y al igual que los pacientes con XLH, los ratones tienen una concentración de calcitriol inapropiadamente normal y se ha demostrado que los ratones Hyp tienen disminución de la 1 alfa hidroxilasa renal comparado con el de ratones normales con el mismo nivel de hipofosfatemia21. Aparte de estos dos defectos el ratón Hyp tienen un defecto primario en los osteoblastos que les impide una mineralización normal de la matriz ósea22. Se ha visto que el defecto de transporte renal de fosfato no se debe a un defecto primariamente renal, sino a la presencia de un factor humoral que inhibe la reabsorción de fosfato y que es inadecuadamente modificado por el osteoblasto anormal del ratón Hyp23.
El gen del XLH fue localizado en el locus Xp22.1 y el gen anormal codifica una proteína de 749 aminoácidos homóloga a los miembros de una familia M13 de metaloendopeptidasas asociadas a la membrana, entre las que se incluye a la endopeptidasa neutra (NEP), la enzima conversora de endotelina (ECE-1) y el antígeno Kell24. Este gen fue denominado originalmente PEX (Phosphate regulating gene with homologies to Endopeptidase on the X Chromosome). El nombre fue posteriormente cambiado a PHEX para evitar confusión con los genes responsables de trastornos de los peroxisomas. El gen PHEX se expresa predominantemente en el hueso y los dientes, y su manifestación en general es de baja magnitud25. La mutación inactivadora del gen PHEX produciría una PHEX endopeptidasa inefectiva que produciría una inadecuada degradación/inactivación de un factor fosfatúrico como la fosfatonina. Las cantidades excesivas circulantes de esta hormona producirían represión de la expresión genética del cotransportador NPT2, con la consiguiente pérdida renal de fosfato e hipofosfatemia.
RAQUITISMO HIPOFOSFATÉMICO AUTOSOMICO DOMINANTE
El raquitismo hipofosfatémico autosómico dominante (ADHR) es otro trastorno hereditario de pérdida aislada renal de fosfato. Su prevalencia es desconocida, pero es menos frecuente que el XLH.
Aproximadamente el 80% de los casos familiares de pérdida renal de fosfato se deben a mutaciones del gen PHEX, pero menos del 50% de los casos esporádicos tienen mutaciones en ese gen26. Existe otro trastorno conocido como enfermedad ósea hipofosfatémica que puede ser una forma frustrada de ADHR.
Los individuos afectados de ADHR tienen una pérdida renal aislada de fósforo con concentraciones de calcitriol sérico inapropiadamente normales27. El ADHR presenta una penetración variable e incompleta. En general, los individuos afectados se presentan en dos subgrupos. Un subgrupo consiste en pacientes con pérdida de fosfato, como adolescentes o adultos. Estos individuos se quejan de dolor óseo, debilidad, fatiga y fracturas, pero no tienen deformidades en las extremidades inferiores; en general, estos individuos son del sexo femenino. El segundo subgrupo consiste en individuos que se presentan durante la niñez con pérdida de fosfato, raquitismo y deformidad de las extremidades inferiores con un patrón similar a la clásica presentación del XHR. En algunos miembros de este subgrupo que presentan raquitismo y pérdida de fosfato durante la niñez, la pérdida urinaria de fosfato desaparece al llegar a la pubertad. A pesar de que la patogénesis del ADHR no se conoce y no hay modelo animal de este trastorno, estudios de ligazón en una gran familia de ADHR han permitido localizar el locus genético de esta enfermedad en el cromosoma 12p1328.
RAQUITISMO HIPOFOSFATÉMICO HEREDITARIO CON HIPERCALCIURIA
El raquitismo hipofosfatémico hereditario con hipercalciuria (HHRH) se trata de una enfermedad autosómica recesiva que se caracteriza por presentar hipofosfatemia, pérdida renal de fosfato con reducción del TmP/GFR, hipercalciuria, niveles plasmáticos elevados de calcitriol, niveles plasmáticos bajos de hormona paratiroidea y elevación de la fosfatasa alcalina29,30.
Desde el punto de vista clínico los pacientes presentan retardo en el crecimiento, dolor óseo, debilidad muscular, deformidad de las extremidades inferiores y evidencia radiológica e histomorfométrica de raquitismo y osteomalacia. Los individuos con alteración de los valores de laboratorio pero sin enfermedad ósea, se presume que son heterocigotos para la enfermedad. La base molecular que subyace al defecto de reabsorción renal de fosfato en el HHRH aún no se ha encontrado. Debido a la ausencia de trastorno en el metabolismo de la vitamina D y debido a que los suplementos de fosfato corrigen totalmente las lesiones óseas en el HHRH, se ha sugerido que en esta enfermedad hay un defecto primario en el transporte renal de fosfato. Estudios de mutagénesis puntual en el transportador npt2 del ratón han producido un fenotipo que es similar al de los pacientes con HHRH.
VALORACION DE UN PACIENTE CON HIPOFOSFATEMIA
La tasa de excreción urinaria de un mineral multiplicada por la filtración glomerular, esta última usualmente estimada a partir de las concentraciones séricas y urinarias de creatinina, puede ser evaluada en relación a las concentraciones séricas de ese mineral, para valorar el papel del riñón a la hora de determinar las concentraciones anormalmente bajas o altas de un determinado mineral en plasma. Por ejemplo, las tasas normales de excreción de fosfato urinario en relación a la tasa de filtración gomerular a pesar de hipofosfatemia plasmática se observa en pacientes con raquitismo hipofosfatémico ligado al X, indicando una pérdida renal de fosfato.
La excreción urinaria de una sustancia como el fosfato por unidad de tasa de filtración glomerular (TFG) se describe por la siguiente expresión:
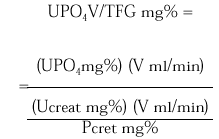
La medición de UPO4 V/TFG en una muestra de orina al azar obtenida en ayunas en relación a la concentración de fosfato sérico puede ser útil como valoración inicial de pacientes que exhiben hipofosfatemia. Los pacientes con hipofosfatemia como consecuencia de una deprivación dietética de fosfato o pérdidas por diarrea de fosfato se esperaría que presentaran máxima conservación renal de fosfato, y por lo tanto un UPO4 V/TFG < 0,03-0,06 mg/100 ml TFG. En contraste, los pacientes que están hipofosfatémicos como consecuencia de una inhibición de la reabsorción tubular de fosfato exhiben tasas urinarias de excreción de fosfato normales o casi normales en estado de equilibrio y un UPO4 V/TFG en el rango normal 0,18-0,78 mg/100 ml TFG. Tales pacientes incluyen a los pacientes con hiperparatiroidismo primario, los pacientes con trastornos hipofosfatémicos hereditarios (raquitismo hipofosfatémico ligado al sexo, raquitismo hipofosfatémico autosómico dominante y raquitismo hipofosfatémico familiar con hipercalciuria) o hipofosfatemia adquirida, como la hipofosfatemia oncogénica, el síndrome de Cushing, la terapia crónica con glucocorticoides.
NOTICIAS
GRUPO DE INVESTIGACIÓN EN ULTRASONIDOS Y METABOLISMO ÓSEO (GIUMO)
El GIUMO es el Grupo de Investigación en Ultrasonidos y Metabolismo Óseo. Fue creado en 1999 en el seno de la Sociedad Española de Investigación en Osteoporosis y Metabolismo Mineral (SEIOMM), como consecuencia de su política de creación de diversos grupos de trabajo y con la finalidad de fomentar y canalizar diferentes líneas de investigación.
Su objetivo principal es el de desarrollar una línea de investigación entre clínicos españoles, aplicando los sistemas de ultrasonidos cuantitativos (QUS) al campo genérico del metabolismo mineral óseo y más concretamente al de la osteoporosis, basado en una metodología rigurosa y seria que permita establecer cuál es el papel que los ultrasonidos pueden jugar en dicho campo. El ultrasonógrafo elegido fue el Sahara (Hologic) que realiza sus determinaciones en el calcáneo utilizando un sistema seco (con gel como conductor).
En un primer estudio efectuado entre 1999 y 2000 y recientemente concluido; en él, se han elaborado las tablas de normalidad de los diferentes parámetros que estima el ultrasonógrafo (BUA, SOS, QUI, Densidad Mineral Ósea [DMO] estimada) en la población española de ambos sexos, de entre 20 y 90 años. Asimismo se calculó la precisión in vivo, in vitro y el coeficiente de variación intermáquinas.
Obviamente, se trata de un grupo abierto a futuras incorporaciones y proyectos de investigación.
Componen el GIUMO:
