





Se han hecho innumerables intentos para lograr una definición que incluya toda la semiología y fisiología que envuelve la epilepsia, y tratar de hacerlo ha sido una tarea que ha tomado muchos años. Aunado a ello se ha propuesto una clasificación en diferentes grupos según su inicio y desencadenamiento, como lo son focales y generalizadas. La presente revisión versará acerca de la epilepsia occipital, clasificada antes dentro de las crisis parciales, en las que se han identificado un curso y un pronóstico favorables en el transcurso del tiempo, y que tienen en común un inicio en la edad temprana. Ha sido descrito que algunos de estos trastornos pudieran tener una base genética hereditaria o bien esporádica en otros, lo cual requiere una investigación exhaustiva. Por lo anterior se hace necesaria una revisión completa acerca de la epilepsia occipital, con el fin de dar claridad conceptual, clínica y diagnóstica de este desorden, así como para hacer aportes al conocimiento de la misma en pro de mejorar la calidad de vida y el tratamiento en la práctica clínica de pacientes que se encuentren dentro del espectro de esta enfermedad.
Epilepsy and its overall semiology and physiology have been subject to Innumerable definition attempts which have taken many years of effort. Likewise, a proposal for an epilepsy classification based on onset and precipitating factors was developed dividing seizures into focal and generalized seizures. This article reviews occipital lobe epilepsy which was formerly classified in the partial seizures group characterized by a favorable course and prognosis over time and by onset during early childhood. A possible genetic hereditary origin and that they may be sporadic has been described for some of these problems and still require a thorough investigation. Thus, it is necessary to conduct an extensive review on occipital lobe epilepsy for obtaining conceptual, clinical and diagnostic clarity as well as, to enhance knowledge on this illness for improving quality of life and clinical treatment in patients experiencing this spectrum of disorders.
La búsqueda y obtención de información a partir de las diferentes bases de datos que en la actualidad están a la vanguardia en temas neurológicos requiere una gran responsabilidad, siendo importante la reunión de la mayor cantidad de datos acerca de temas específicos, como es el caso de la epilepsia occipital, que a edades tempranas de la vida es muy común, mientras que en el adulto ocurre con menor frecuencia. Por tanto, siendo una entidad de la que poco se habla, pero que está dentro de la práctica clínica diaria, se hace necesario diferenciar las entidades más frecuentes, analizándolas desde la perspectiva de aquellas cuya etiología es conocida, de las que en realidad se encuentran dentro de un grupo idiopático.
Es así como en las primeras se describen lesiones estructurales específicas del sistema nervioso central. Por otro lado, las que se hallan en la clasificación de origen indeterminado se pueden dividir por edad de inicio, configurando una temprana, una tardía y por supuesto otra de edad intermedia. Históricamente, la primera descripción formal de epilepsia occipital fue realizada por Gastaut en 1982, cuando reunió una cantidad considerable de pacientes y describió en ellos una afección en la que podía encontrarse buen control de las crisis y características electroencefalográficas en todos los casos. En 1989, Panayiotopoulos describió una serie de casos con mayor amplitud en cuanto a sintomatología y semiología que el anterior investigador. Gracias a estas contribuciones, la Liga Internacional Contra la Epilepsia (ILAE) en 1999 la empieza a clasificar en una categoría por aparte.
Materiales y métodosSe llevó a cabo una revisión de diferentes bases de datos: PubMed, Lilacs, Cochrane y Ovid, de donde se obtuvieron en total 16 artículos, 3 de ellos de casos y controles, 2 de reporte de caso y 11 de revisión de tema, de los cuales se obtuvo la mayor parte de la información para el presente artículo acerca de la epilepsia occipital. No fueron usados criterios de exclusión para ninguno de ellos. De algunos se tomaron tablas e imágenes para ilustrar de manera clara y concreta algunos diagnósticos diferenciales y características propias de la epilepsia.
EtiologíaLa mayoría de veces no se encuentra un sustrato etiológico específico para la producción de un foco epileptogénico más allá de la lesión propia o anormalidades congénitas o genéticas cerebrales que pueden explicar las crisis1. Estudios de cohorte en centros de referencia han sugerido que de las epilepsias parciales al menos el 25% son estructurales, el 18% de etiología indeterminada y el 0,1% de origen genético2. Dentro de las etiologías más comunes se encuentran los tumores, las malformaciones vasculares, las congénitas y las alteraciones asociadas con una lesión antigua. En algunos países se ha determinado la neurocisticercosis como causa importante de crisis epilépticas. Con el antecedente de lesión es muy probable que no sea efectivo el tratamiento establecido, es decir, que la etiología estaría relacionada en forma directa con el control de las crisis2-4.
Cuadro clínico de las epilepsias occipitalesCabe mencionar el cuadro clínico general en la presentación de la epilepsia occipital. Entre las características importantes están las crisis que por lo regular ocurren en estado de vigilia, y cuando son diurnas van asociadas con sintomatología visual, como escotomas, amaurosis, fosfenos o alteraciones en un hemicampo o en todo el campo visual. Se acompañan de cefalea de inicio súbito en el periodo postictal, de características migrañosas e incluso podría presentar vómito5. También hay supraversión de la mirada, hipertonía generalizada y cianosis (tabla 1). Toda la sintomatología por lo regular no tiene una duración mayor de un minuto5,6.
Características clínicas de la epilepsia occipital
| Características clínicas de la epilepsia occipital |
| Vómito ictal |
| Desviación tónica oculocefálica |
| Episodios migrañosos |
| Crisis visuales |
| Crisis secundariamente generalizas |
| Crisis parciales clónicas |
| Ausencias típicas |
| Anartria y crisis motoras hemifaciales |
| Crisis en vigilia |
| Crisis durante el sueño |
| Crisis prolongadas |
| Status |
Fuente: tomado de Caraballo et al.1.
Este tipo se encuentra dentro de las epilepsias focales de la infancia; es más frecuente en edad preescolar, teniendo su pico de inicio a los 2 años. En una serie de 74 casos, Caraballo1 describe un antecedente familiar importante, obteniendo un 30% de asociación entre las características del evento, dentro de las cuales se encontraban agitación, versión cefálica y ocular, además de compromiso del estado de conciencia, vómito ictal, sialorrea, palidez y diaforesis, teniendo como explicación un foco convulsivo debajo de la cisura calcarina con propagación a estructuras límbicas (tabla 2)5,7.
Tipos de epilepsias occipitales
| Epilepsias occipitales |
|---|
| Idiopáticas |
| No están relacionadas con anormalidades cerebrales estructurales: |
| Epilepsia occipital benigna temprana (Panayiotopoulos). |
| Epilepsia occipital benigna tardía (Gastaut) |
| Epilepsia occipital fotosensitiva |
| Epilepsia parcial benigna atípica de la infancia o pseudo Lennox-Gastaut |
| Epilepsia rolándica |
| Sintomáticas |
| Asociadas con malformaciones del desarrollo cortical: |
| Displasia cortical occipital |
| Heterotopia periventricular occipital |
| Polimicrogiria |
| Epilepsias mioclónicas progresivas |
| Enfermedades mitocondriales |
Fuente: tomado de Carrizosa Moog y Castaño Parra2.
Panayiotopoulos, un eminente neurólogo y neurofisiólogo nacido en Tinos en 1938, escogió, de una muestra de 900 pacientes, 24 niños con la particularidad que 21 de ellos eran varones y solo 3 tenían epilepsias sintomáticas2. En la mitad de los casos los ataques duraban >1h. Los EEG de los 21 casos analizados revelaron grandes variaciones, con una distribución de la siguiente manera: 12 tenían presencia de paroxismos occipitales, descargas aisladas u ondas agudas extraoccipitales, 2 con actividad central, 2 en línea media, uno frontal, a uno se le documentaron descargas generalizadas y 3 tenían EEG normal. Así, la atención fue centrada en la parte gruesa del grupo en la cual había espigas occipitales, las cuales se clasificaron como epilepsias benignas de la infancia7-9.
De esta manera, Panayiotopoulos estableció unos criterios entre los que mencionaba los episodios con las características ya descritas, edad de inicio a los 2 años, neurodesarrollo sin alteraciones, ninguna anormalidad en las neuroimágenes y EEG con un periodo postictal donde se revela un foco de región occipital alternante con frontal y temporal, indicando una migración en el foco epileptogénico, que tiene varias teorías como la migración por maduración del sistema nervioso y la fisiopatología compartida con la crisis rolándica, e indicando como característica desencadenante la supresión de la visión central7,8.
Es indudable que el vómito ictal debe ser criterio mayor para el diagnóstico, puesto que el cuadro lo incluyó siempre como síntoma cardinal con la desviación oculocefálica y en el 40% de los casos hubo presencia de convulsiones febriles (tabla 3)8,10.
Comparación entre epilepsias tipo Panayiotopoulos y tipo Gastaut
| Epilepsia occipital tipo Panayiotopoulos | Epilepsia occipital tipo Gastaut |
|---|---|
| Crisis prolongadas (de 5-30min a horas) | Crisis cortas (segundos a menos de 1-3min) |
| De preferencias nocturnas | Diurnas |
| Poco numerosas (1-5 en toda la vida) | Frecuentes (a menudo diarias) |
| Ocurren en un intervalo corto (un año) | Se presentan durante 2-3 o más años |
| Muy buen pronóstico | Pronóstico reservado |
| Desviación ocular | Desviación ocular |
| No alucinaciones visuales (o muy raras) | Alucinaciones visuales |
| EEG crítico: actividad lenta rítmica, monomorfa, mezclada con puntas de inicio posterior | EEG crítico: actividad rápida occipital |
Fuente: tomado de Palencia-Luaces3.
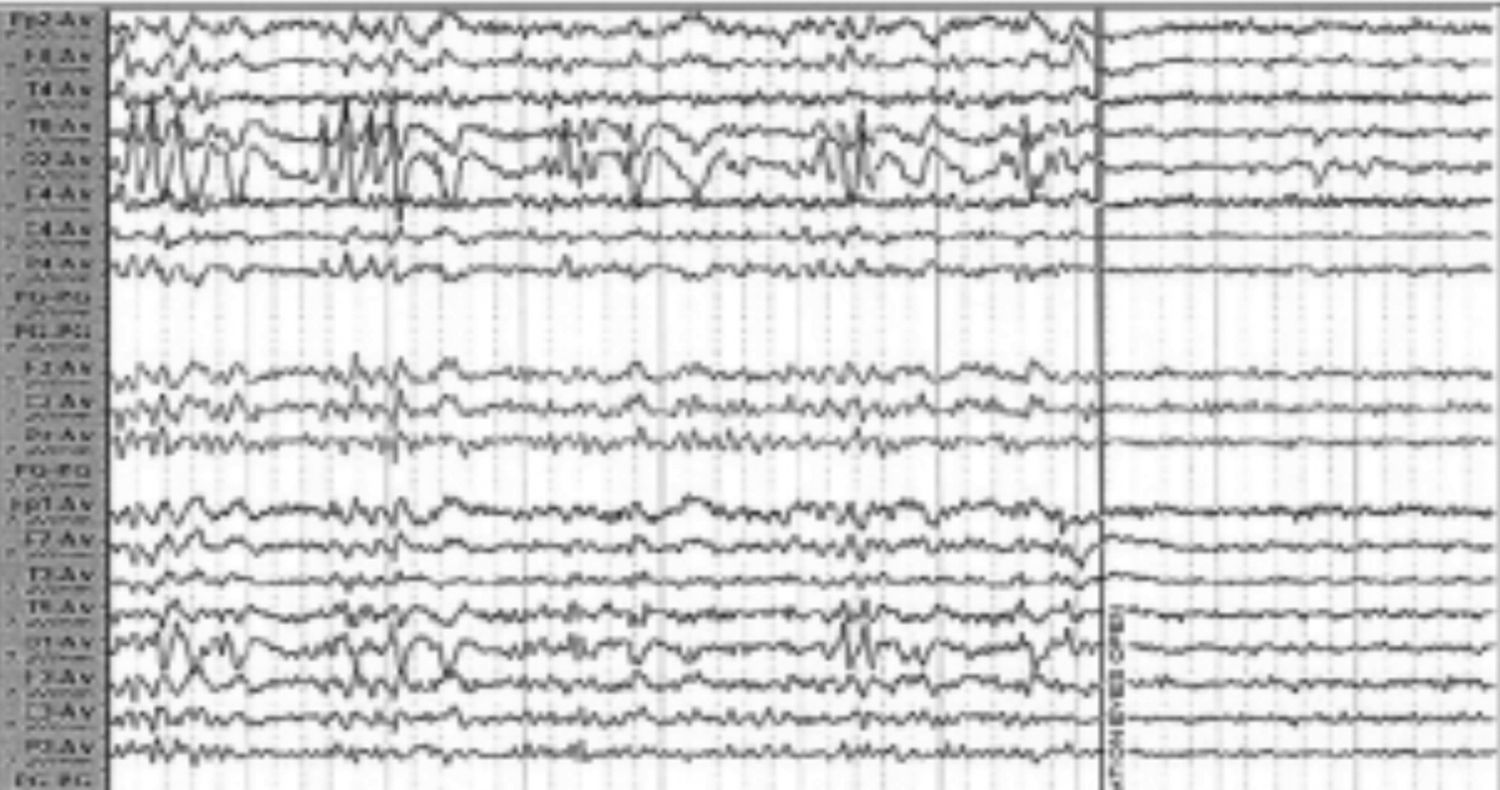
Este tipo de epilepsia tiene su inicio entre los 6 y 8 años, con un antecedente familiar importante hasta en el 40%; la mayoría de veces se encuentra amaurosis o discromatopsias, además de ilusiones ópticas, con una duración <3min12. El síntoma postictal más frecuente es la cefalea. En el EEG se aprecian puntas de onda, pero solo con los párpados cerrados (fig. 1)2.

Espigas en región occipital y a la derecha, la supresión de las mismas con la mirada.
Fuente: tomado de Carrizosa Moog y Castaño Parra2.
Entre el 30 y el 40% cursan con cefalea poscrisis, lo cual implica hacer el diagnóstico diferencial con migraña basilar y migraña con aura7. Se encuentra descrito en la literatura un patrón autosómico dominante con penetrancia variable y hasta el 26% de los familiares pueden presentar anomalías en el EEG8,11,12.
Se podría mencionar además la epilepsia infantil con punta occipital inducida o epilepsia occipital idiopática fotosensitiva5. Aunque con frecuencia se resta importancia clínica a los estímulos externos, en algunos pacientes con epilepsia de base se puede comprobar esta afección, que por lo regular se inicia desde los 5 años hasta la adolescencia temprana. Las crisis pueden ser facilitadas por estímulos luminosos y cursan con crisis asociadas con discromatopsias, desviación de la mirada, vómito y cefalea13. Como último grupo se encuentra la epilepsia parcial benigna atípica de la infancia o síndrome pseudo Lennox-Gastaut, que tiene una edad de inicio entre los 2 y 6 años, cursa con crisis de ausencia, además de crisis rolándicas, asociado con alteración cognitiva6,14.
Aicardi y Newton en 1987 estudiaron 21 pacientes con paroxismos occipitales reactivos a la apertura palpebral (7 casos con un curso benigno y 3 tuvieron epilepsia), de los cuales 6 presentaron alteración cognitiva grave8,15,16.
Epilepsia occipital de presentación tardíaLoiseau17 en 1992 analizó 108 pacientes con convulsiones en la adolescencia, con un rango de edad entre los 13 y 18 años y predominio del género masculino. La define como una afección transitoria caracterizada por crisis motoras parciales simples, a menudo con generalización secundaria, únicas o múltiples, con predominancia en la vigilia y de curso benigno9. En exámenes como el EEG reporta normalidad y tampoco existen antecedentes familiares importantes de epilepsia. En un artículo del Acta de Neurocirugía, Nanying describe un estudio con 125 pacientes de entre 10 y 18 años y seguimiento a 7 años, con predominancia de crisis focales motoras, con clonías o versión cefálica, siendo excepcional la aparición después de los 12 años. El espectro de síntomas es muy parecido a los descritos antes en las epilepsias en niños, es decir, auras o alucinaciones visuales, amaurosis, supraversión de la mirada, además de sensación de movimiento de los ojos y del párpado. En todos se desencadenó migraña de intensidad moderada10.
En el EEG se ven complejos punta-onda o elementos agudos amplios que continúan rítmicos a regiones temporales posteriores, hallazgos que se ven más cuando el paciente tiene los párpados cerrados. Las descargas occipitales pueden ser propagadas a la región temporal14. La dificultad para hacer el diagnóstico de epilepsia occipital en un adulto no radica entonces en la etiología, ni en la diferenciación de síntomas y es probable que tampoco en la edad de aparición —aunque esporádicamente aparece en adultos—, sino que sería en estos casos la propagación a otras zonas, lo cual podría dar sintomatología complementaria que distorsiona el diagnóstico principal. Por ejemplo, las crisis son de características parciales complejas9,18.
Epilepsia de la infancia con puntas centrotemporales (epilepsia rolándica)Fue descrita en 1952 por primera vez y está incluida dentro de las epilepsias idiopáticas. La ILAE la define como un síndrome de crisis parciales, simples, motoras, de localización hemifacial y que puede estar asociado con síntomas somatosensoriales, con tendencia a las crisis tónico-clónicas generalizadas.
Su incidencia se encuentra alrededor de 5 a 20 casos por cada 100.000 menores de 15 años7.
Para hablar un poco de cifras cabe mencionar que hasta en el 50% de los niños que se ven afectados por este trastorno existen antecedentes de crisis febriles; aunado a esto, las alteraciones funcionales de la maduración de la corteza junto con factores externos de la maduración contribuirían a la conversión sintomática, designándolo susceptibilidad a presentar epilepsia benigna11,12,18.
Las crisis suelen ser parciales, ocurren casi siempre durante el sueño, aunque también pueden presentarse despiertos, es frecuente la desviación de la mirada hacia un lado, con tendencia a la evolución a hemiconvulsiones de cara y extremidades, terminando en crisis tónico-clónicas generalizadas4. Genéticamente puede presumirse un patrón autosómico dominante con una penetrancia variable, gracias a estudios en gemelos monocigotos con descargas rolándicas5. La teoría genética ha sido comprobada como con una alteración en la región 15q14, la misma afectada en la epilepsia mioclónica juvenil (tabla 3)6,7.
El hallazgo del paroxismo rolándico en el EEG es fundamental para establecer el diagnóstico, aunque su localización es variable. En estudios imagenológicos como TAC y RM no suelen verse alteraciones, aunque se han descrito algunas que tienen que ver con entidades como la macrogiria opercular, la displasia cortical, las lesiones postraumáticas y las asimetrías con alteraciones de sustancia blanca19.
Las descargas paroxísticas antes descritas pueden manifestarse en la clínica de muchas maneras, ya sea como fenómenos visuales ictales o crisis de semiología diferente, aunque también pueden ocurrir de manera asintomática con tendencia a la desaparición con la edad, lo que no configura un espectro de lesión estructural19.
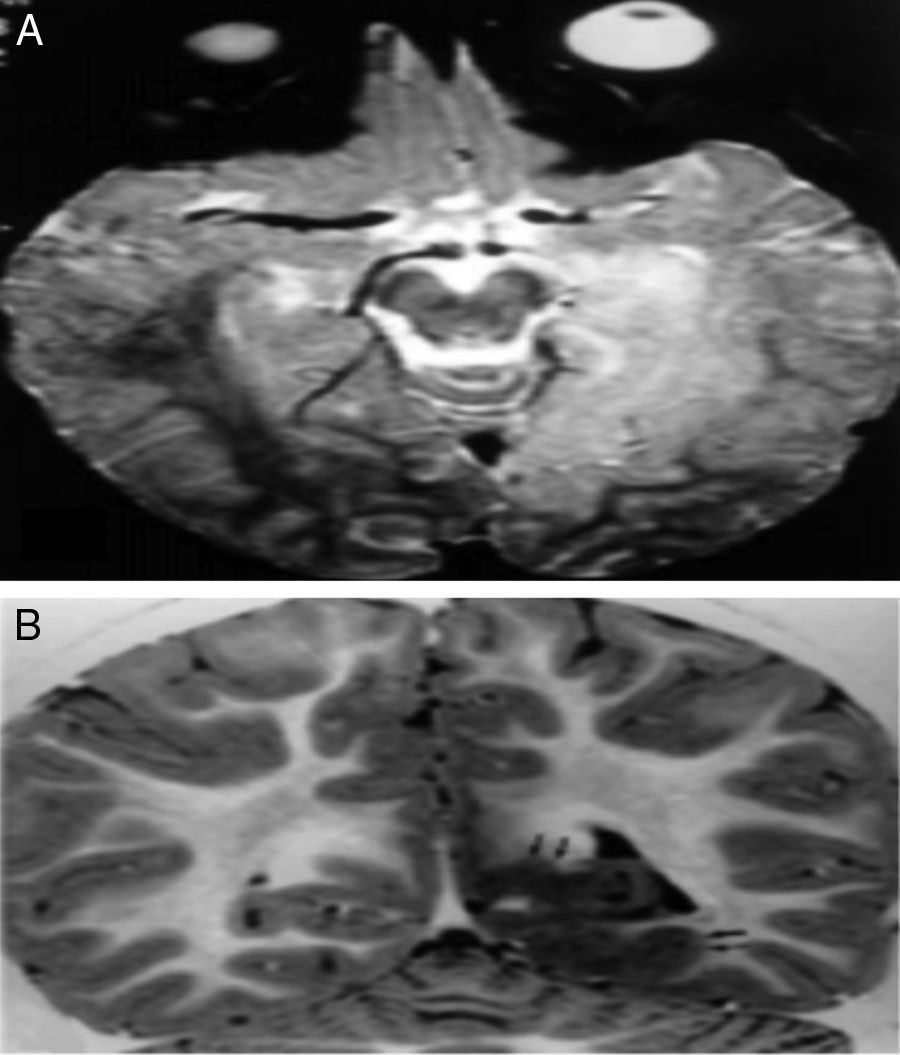
Epilepsias occipitales sintomáticasMalformaciones del desarrollo corticalDisplasia cortical occipitalDe inicio temprano, manifestado con episodios de epilepsia que no cede al manejo con medicamentos. En la resonancia los hallazgos van desde mala delineación de la corteza hasta engrosamiento focal. Solo hay respuesta al manejo quirúrgico (fig. 2)4.
 IRM con extensa lesión temporo-occipito-basal hiperintensa en T2. B) Serie de inversión recuperación con engrosamiento de la sustancia gris y borramiento de la unión córtico-subcórtica. Fuente: tomado de Taylor et al.9.")
Displasia cortical occipital. A) IRM con extensa lesión temporo-occipito-basal hiperintensa en T2. B) Serie de inversión recuperación con engrosamiento de la sustancia gris y borramiento de la unión córtico-subcórtica.
Fuente: tomado de Taylor et al.9.
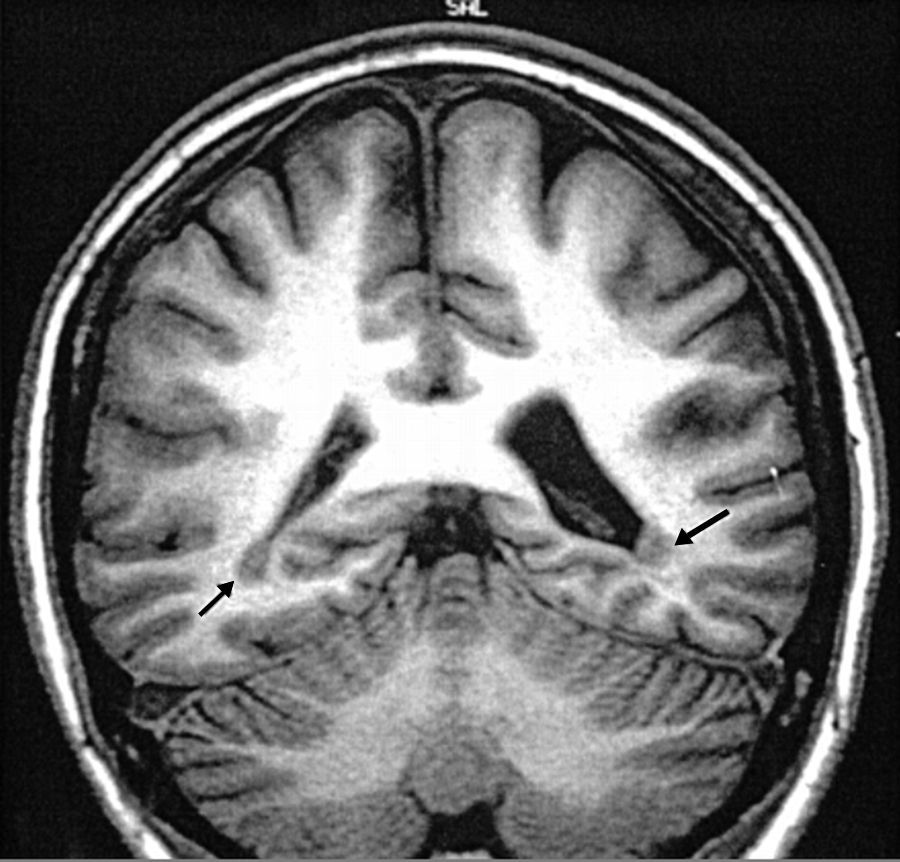
Es una ubicación errada de la sustancia gris dentro del parénquima cerebral. Se ha observado una asociación con el gen de la filamin-1 y puede iniciarse desde los 20 años. En las imágenes se pueden ver lesiones isointensas a la sustancia gris, de bordes circunscritos, aunque la zona epileptogénica es más grande que la de isointensidad (fig. 3)4.
. Fuente: tomado de Taylor et al.9.")
Heterotopia periventricular occipital. T 1: imagen coronal demostrando heterotopia periventricular bioccipital (flechas).
Fuente: tomado de Taylor et al.9.
Se asocia con anormalidades en el cromosoma 17; las imágenes muestran el signo de doble corteza donde se aprecia una banda de sustancia gris paralela a la corteza8.
PolimicrogiriaEn este tipo de epilepsia existe un exceso de surcos en la corteza cerebral, dando como resultado giros pequeños. La causa es la alteración en la organización cortical posmigratoria y puede aparecer entre los 10 y 20 años8.
Enfermedades mitocondrialesSe caracterizan por alteraciones primarias del metabolismo oxidativo con compromiso sistémico, como sucede en la llamada enfermedad Melas (miopatía mitocondrial encefalopática, acidosis láctica y episodios tipo ictus). Cursan con episodios de vómito, migraña y convulsiones8.
ConclusiónCon esta revisión se aprecia el espectro de la epilepsia occipital, evidenciando algunos diagnósticos diferenciales que deben tenerse en cuenta en la práctica clínica cuando se sospecha una entidad de este tipo. Se destacan varios puntos: en cuanto a la edad de inicio, por lo general ocurre en edades tempranas de la vida y su pronóstico es bueno porque tiende a resolverse en el mismo periodo de tiempo en el que se presenta. La base genética se encuentra aún en investigación, pero se ha podido vislumbrar que hay una clara asociación con algunas lesiones genéticas como la epilepsia juvenil. En general es una enfermedad tratable con gran variedad de arsenal terapéutico, por la edad en la que se presenta, aunque con algunas excepciones en las que existen daños estructurales, pues es bien sabido que el tejido cerebral no posee la misma capacidad de regenerar células y no dejar secuelas como sucede en otros tejidos. Es indudable que una buena historia clínica y una anamnesis detallada, en donde se relaten cronológicamente el inicio y la duración de los síntomas, constituyen la base para establecer un diagnóstico preciso, brindar un tratamiento eficaz y sobre todo mejorar la calidad de vida.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.