


La transposición penoescrotal es una anomalía congénita donde el escroto se localiza en posición superior y anterior al pene. Se puede clasificar según su compromiso en transposición completa o incompleta, siendo más frecuente esta última. Se han reportado alrededor de 27 casos en la literatura de transposición penoescrotal completa y la gran mayoría se ha asociado con otras malformaciones congénitas. Presentamos dos casos de óbitos fetales con transposición penoescrotal completa con malformaciones congénitas, y además realizamos una revisión de la literatura de los casos publicados.
Penoscrotal transposition is a congenital anomaly in which the scrotum is positioned superiorly and anteriorly to the penis. It may be classified as complete or incomplete transposition, according to severity. Incomplete type is more common. Around 27 cases of complete penoscrotal transposition have been reported in the literature usually associated with other congenital malformations. We report on two cases on fetal obitus with complete penoscrotal transposition associated with congenital malformations. We also reviewed the literature on published cases.
La transposición penoescrotal (TPE) es una malformación congénita poco frecuente, de etiología desconocida, donde el escroto se encuentra en localización superior y anterior al pene1. Se puede clasificar según su compromiso, bien sea transposición completa o incompleta2. La mayoría de los casos reportados son esporádicos3, pero en los casos familiares se puede encontrar un patrón genético4. Debido a su baja frecuencia presentamos dos casos de óbitos fetales con TPE completa con malformaciones congénitas asociadas con esta entidad. Se realiza una revisión de la literatura de los casos publicados.
Presentación de casosCaso 1Feto masculino de 23 semanas por antropometría fetal, producto de madre primigestante de 21 años, con paraclínicos prenatales de ecografía fetal con evidencia de agenesia renal y anhidramnios. Se realiza cesárea y se obtiene producto único de sexo masculino con APGAR 0-0-0. Líquido ausente y membranas ovulares adheridas. El resto de paraclínicos sin alteraciones.
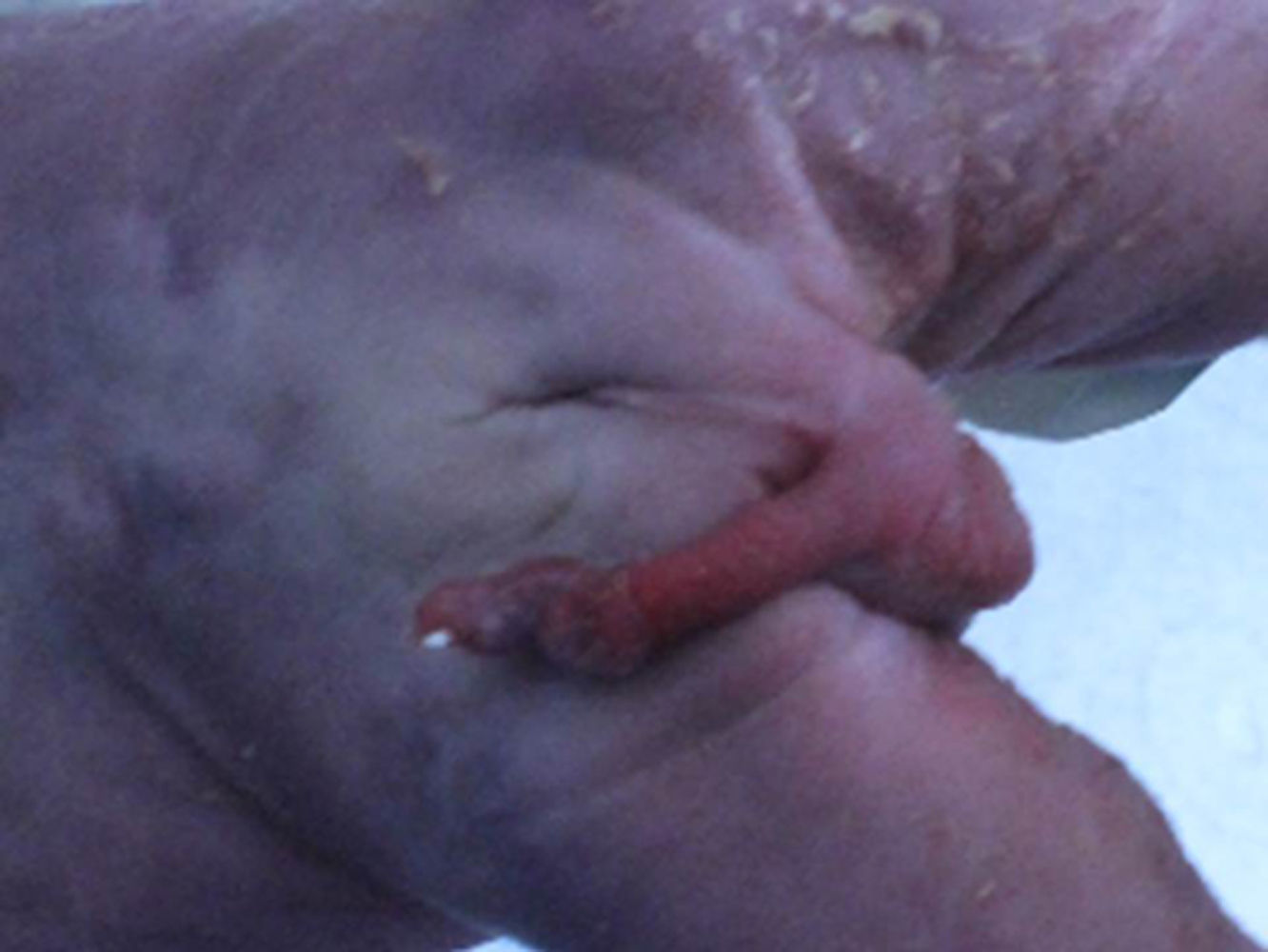
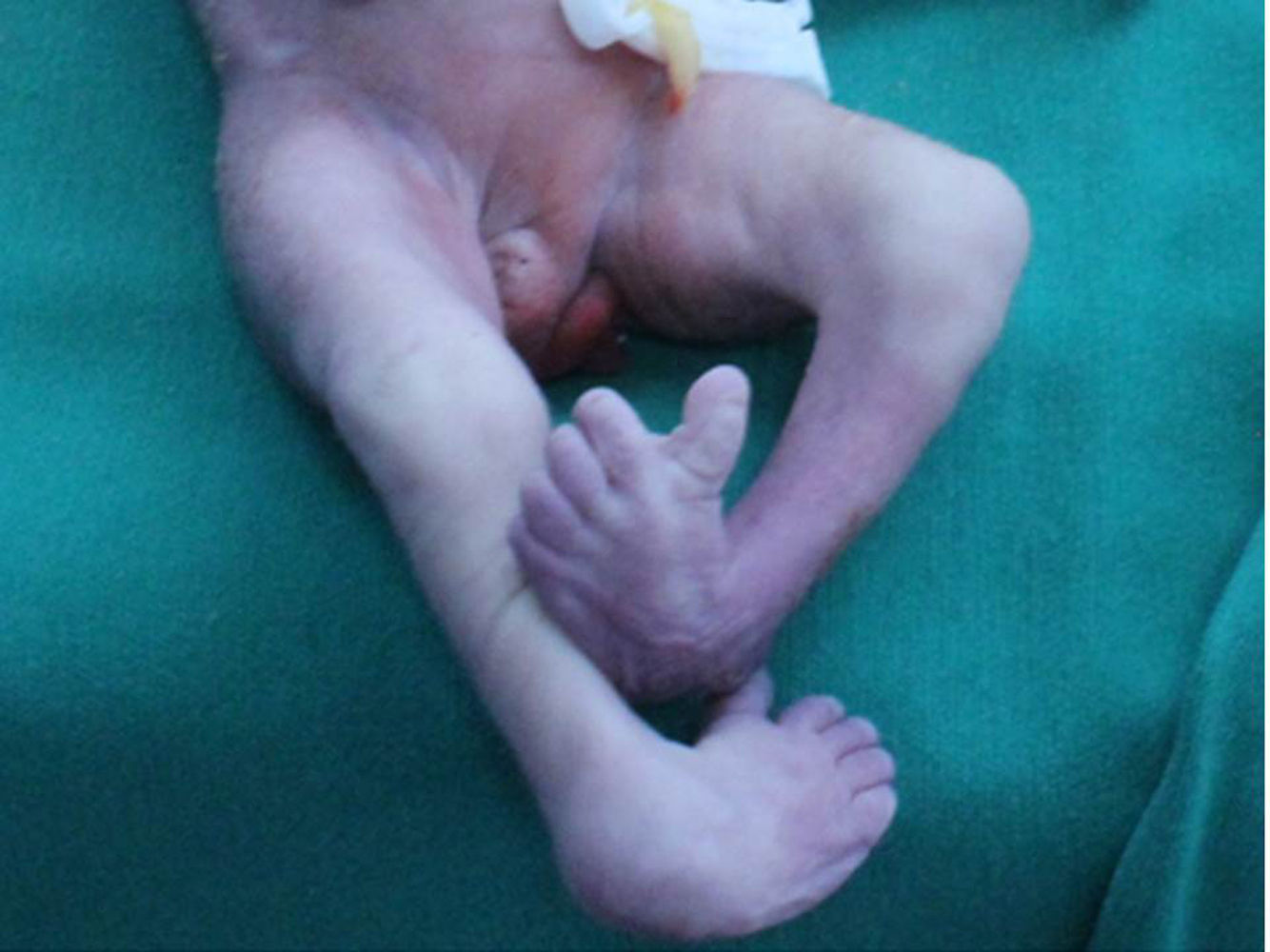
En el examen macroscópico se observa feto con TPE completa (fig. 1), ano imperforado, arteria umbilical única y pie equino varo (fig. 2). Presenta además, alteración del sistema urinario por agenesia bilateral de uréteres, riñones y de vejiga. En el estudio anatomopatológico no se observó presencia de tejido renal.


Feto gemelar masculino de 33 semanas, producto de segunda gestación caracterizada por restricción selectiva del feto N2 (caso de estudio); madre de 25 años con antecedente de granulomatosis con poliangitis (Wegener), queratoconjuntivitis e hipertiroidismo. Se documenta durante el embarazo oligohidramnios y una diferencia del 30% del peso con el feto N1. Realizan estudio prenatal con FISH y cariotipo de ambos fetos, con resultados 46XY. Se realiza cesárea de urgencia y se obtiene producto de embarazo gemelar biamniótico. El recién nacido N2 inicia con dificultad respiratoria por lo que requiere intubación orotraqueal y le trasladan a UCIN, donde fallece al siguiente día de nacido.
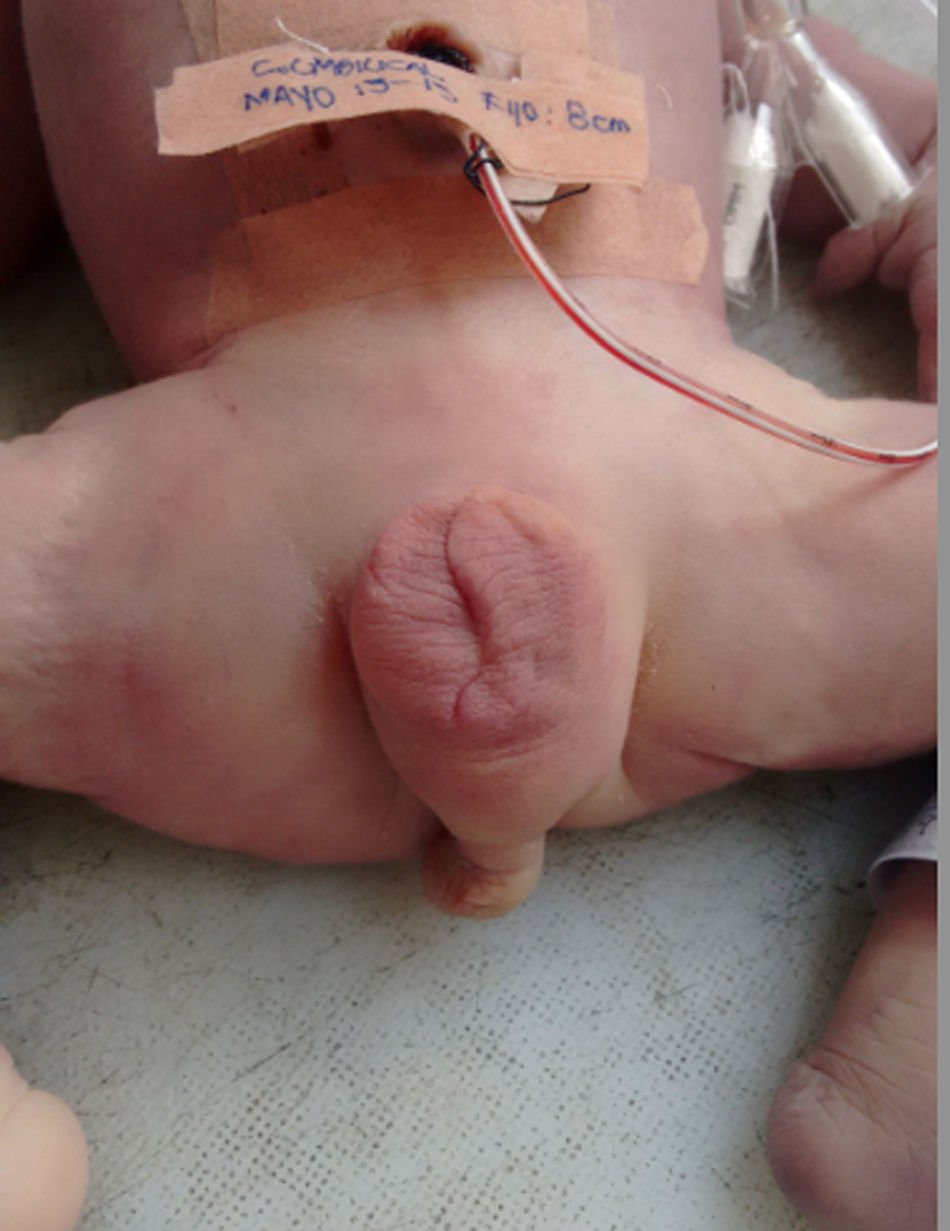
En el examen macroscópico se observa TPE completa (fig. 3), asociada con ano imperforado, arteria umbilical única, hipospadia, agenesia renal derecha, displasia renal multiquística izquierda, hipoplasia pulmonar, cardiomegalia, luxación de cadera, hipoplasia de pulgares, implantación baja de orejas, hipertelorismo y puente nasal deprimido. En el estudio anatomopatológico lo relevante son los cortes de riñón, que muestras un parénquima con pérdida de la arquitectura dado por presencia de lesiones quísticas.
Discusión
La TPE es una anomalía poco común en los recién nacidos1. La primera descripción patológica se remonta a 1923 por Appleby5. La clasificación depende del posicionamiento anatómico del pene y escroto, siendo mayor o menor2. La TPE mayor se clasifica en completa, donde el escroto intacto está ubicado sobre el pene que emerge del perineo2,6, o incompleta como en la mayoría de los casos donde el pene se ubica en la mitad del escroto. La transposición menor puede ser bilateral (simétrica) o unilateral1,6.
La etiología se desconoce, algunos autores proponen que la anomalía congénita es resultado de una obstrucción completa del tracto urinario superior a nivel de la unión pelvouretral o del gubernáculo1,2,7,8, la cual genera una migración aberrante de las células escrotales secundaria al mal posicionamiento del tubérculo genital que se da en la semana 4 o 5 de gestación2, o incluso hasta la semana 63. En el proceso antes mencionado la obstrucción también genera hipoplasia secundaria de la vejiga y uréteres, que con la distensión rectal que se crea por la atresia anal, da lugar a una disrupción en la orientación cráneo-caudal del septum urorrectal, interfiriendo con el posicionamiento anatómico normal del tubérculo genital2. Otra teoría sugiere que el tubérculo fálico es intrínsecamente anormal, dando de esta manera afectación del cuerpo, lo que explica el pene anormal y otras anormalidades genitales1.
El estudio genético de la transposición penoescrotal no muestra semejanza en todos los fetos reportados. En la mayoría de los casos familiares se ha encontrado una herencia ligada al cromosoma X, asociada en algunos casos con el síndrome de Simpson-Golabi-Behmel1,2. También se ha asociado con deleciones del cromosoma 132,4 y el síndrome de Klinefelter9. Existe un caso único de herencia recesiva con mutación perdedora de función del gen ZBTB16, ubicado en el cromosoma 13q232.
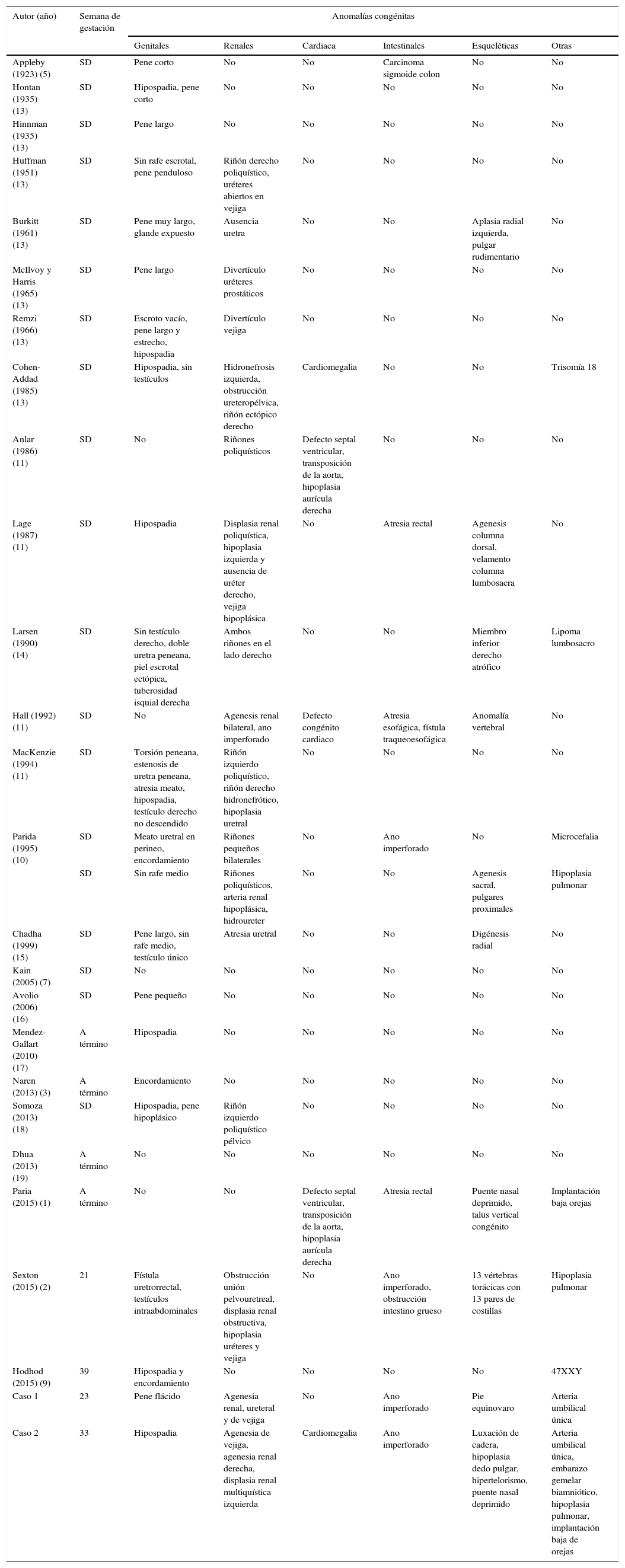
En la literatura médica se han descrito alrededor de 27 reportes de TPE completa (tabla 1)1–20. Los hallazgos que se evidencian son la presencia de anormalidades genitales (22/27), en especial hipospadia (9/22), y extragenitales, tales como alteraciones renales (17/27), siendo la enfermedad poliquística la más frecuente (7/17). Otras malformaciones reportadas son las esqueléticas (10/27), intestinales (8/27), cardiacas (5/27), pulmonares (3/27) y del sistema nervioso central (1/27). Cabe resaltar los dos únicos casos que se presentan con anormalidad cariotípica: una trisomía 18 y un síndrome de Klinefelter.
Casos publicados de transposición penoescrotal completa
| Autor (año) | Semana de gestación | Anomalías congénitas | |||||
|---|---|---|---|---|---|---|---|
| Genitales | Renales | Cardiaca | Intestinales | Esqueléticas | Otras | ||
| Appleby (1923) (5) | SD | Pene corto | No | No | Carcinoma sigmoide colon | No | No |
| Hontan (1935) (13) | SD | Hipospadia, pene corto | No | No | No | No | No |
| Hinnman (1935) (13) | SD | Pene largo | No | No | No | No | No |
| Huffman (1951) (13) | SD | Sin rafe escrotal, pene penduloso | Riñón derecho poliquístico, uréteres abiertos en vejiga | No | No | No | No |
| Burkitt (1961) (13) | SD | Pene muy largo, glande expuesto | Ausencia uretra | No | No | Aplasia radial izquierda, pulgar rudimentario | No |
| McIlvoy y Harris (1965) (13) | SD | Pene largo | Divertículo uréteres prostáticos | No | No | No | No |
| Remzi (1966) (13) | SD | Escroto vacío, pene largo y estrecho, hipospadia | Divertículo vejiga | No | No | No | No |
| Cohen-Addad (1985) (13) | SD | Hipospadia, sin testículos | Hidronefrosis izquierda, obstrucción ureteropélvica, riñón ectópico derecho | Cardiomegalia | No | No | Trisomía 18 |
| Anlar (1986) (11) | SD | No | Riñones poliquísticos | Defecto septal ventricular, transposición de la aorta, hipoplasia aurícula derecha | No | No | No |
| Lage (1987) (11) | SD | Hipospadia | Displasia renal poliquística, hipoplasia izquierda y ausencia de uréter derecho, vejiga hipoplásica | No | Atresia rectal | Agenesis columna dorsal, velamento columna lumbosacra | No |
| Larsen (1990) (14) | SD | Sin testículo derecho, doble uretra peneana, piel escrotal ectópica, tuberosidad isquial derecha | Ambos riñones en el lado derecho | No | No | Miembro inferior derecho atrófico | Lipoma lumbosacro |
| Hall (1992) (11) | SD | No | Agenesis renal bilateral, ano imperforado | Defecto congénito cardiaco | Atresia esofágica, fístula traqueoesofágica | Anomalía vertebral | No |
| MacKenzie (1994) (11) | SD | Torsión peneana, estenosis de uretra peneana, atresia meato, hipospadia, testículo derecho no descendido | Riñón izquierdo poliquístico, riñón derecho hidronefrótico, hipoplasia uretral | No | No | No | No |
| Parida (1995) (10) | SD | Meato uretral en perineo, encordamiento | Riñones pequeños bilaterales | No | Ano imperforado | No | Microcefalia |
| SD | Sin rafe medio | Riñones poliquísticos, arteria renal hipoplásica, hidroureter | No | No | Agenesis sacral, pulgares proximales | Hipoplasia pulmonar | |
| Chadha (1999) (15) | SD | Pene largo, sin rafe medio, testículo único | Atresia uretral | No | No | Digénesis radial | No |
| Kain (2005) (7) | SD | No | No | No | No | No | No |
| Avolio (2006) (16) | SD | Pene pequeño | No | No | No | No | No |
| Mendez-Gallart (2010) (17) | A término | Hipospadia | No | No | No | No | No |
| Naren (2013) (3) | A término | Encordamiento | No | No | No | No | No |
| Somoza (2013) (18) | SD | Hipospadia, pene hipoplásico | Riñón izquierdo poliquístico pélvico | No | No | No | No |
| Dhua (2013) (19) | A término | No | No | No | No | No | No |
| Paria (2015) (1) | A término | No | No | Defecto septal ventricular, transposición de la aorta, hipoplasia aurícula derecha | Atresia rectal | Puente nasal deprimido, talus vertical congénito | Implantación baja orejas |
| Sexton (2015) (2) | 21 | Fístula uretrorrectal, testículos intraabdominales | Obstrucción unión pelvouretreal, displasia renal obstructiva, hipoplasia uréteres y vejiga | No | Ano imperforado, obstrucción intestino grueso | 13 vértebras torácicas con 13 pares de costillas | Hipoplasia pulmonar |
| Hodhod (2015) (9) | 39 | Hipospadia y encordamiento | No | No | No | No | 47XXY |
| Caso 1 | 23 | Pene flácido | Agenesia renal, ureteral y de vejiga | No | Ano imperforado | Pie equinovaro | Arteria umbilical única |
| Caso 2 | 33 | Hipospadia | Agenesia de vejiga, agenesia renal derecha, displasia renal multiquística izquierda | Cardiomegalia | Ano imperforado | Luxación de cadera, hipoplasia dedo pulgar, hipertelorismo, puente nasal deprimido | Arteria umbilical única, embarazo gemelar biamniótico, hipoplasia pulmonar, implantación baja de orejas |
SD: sin determinar
El diagnóstico in útero se puede realizar por ultrasonido, escáner 3D/4D o resonancia magnética fetal2. En los pacientes donde la presentación clínica es compatible con la vida, el tratamiento es quirúrgico. Las complicaciones quirúrgicas son daño testicular y uretral, fístula urinaria y edema del pene20, por tal motivo, se recomienda realizar el procedimiento quirúrgico entre la semana 12 y 18 después del nacimiento, donde el principio básico es el desplazamiento del tejido escrotal posteriormente y la del pene antes20.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.