




Impacto de la farmacogenómica en el proceso de investigación y desarrollo de un nuevo medicamento
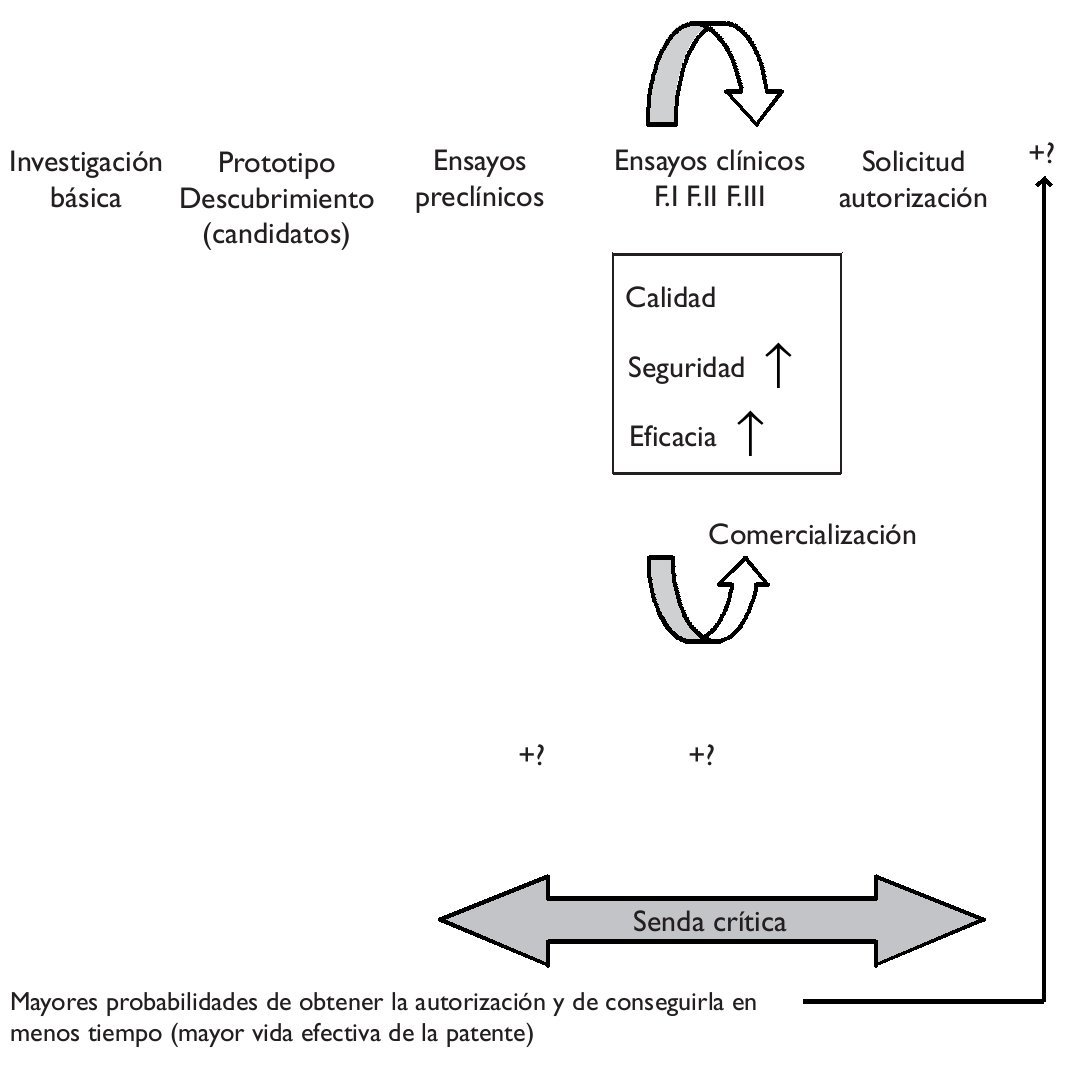
Es sabido que el proceso de investigación y desarrollo de un nuevo medicamento es duradero y muy costoso. En poco tiempo está teniendo lugar un importante incremento de costes que, unido a la mayor duración del proceso y a la obtención de peores resultados en términos de innovación, explican tanto el éxito como el malogro del modelo blockbuster de la industria, es decir, la estrategia de centrarse en (pocos) productos muy rentables, con unas ventas globales superiores a mil millones de dólares anuales. La estrategia blockbuster está dando señales de constituir un modelo ya caduco, precisamente por las crecientes dificultades para obtener este tipo de productos. Ante esta situación, hace unos años, la FDA estadounidense (Food and Drug Administration) emprendió la iniciativa critical path ("la senda crítica"), entendiendo que los avances en las ciencias básicas no estaban siendo debidamente acompasados por los conseguidos en las ciencias aplicadas, ampliando la distancia entre potencialidades y realidades y conduciendo a las autoridades a fijar niveles de exigencia más conservadores, de modo que el proceso hacia la obtención de un nuevo medicamento se tornaba finalmente más difícil1.La iniciativa de "la senda crítica" pretende modernizar el proceso de desarrollo que abarca desde el descubrimiento de un nuevo medicamento hasta su lanzamiento al mercado, obteniendo nuevas herramientas científicas y técnicas que favorezcan un proceso más eficiente que otorgue mayores posibilidades de obtener productos seguros y eficaces en menor tiempo y a menores costes (fig. 1).
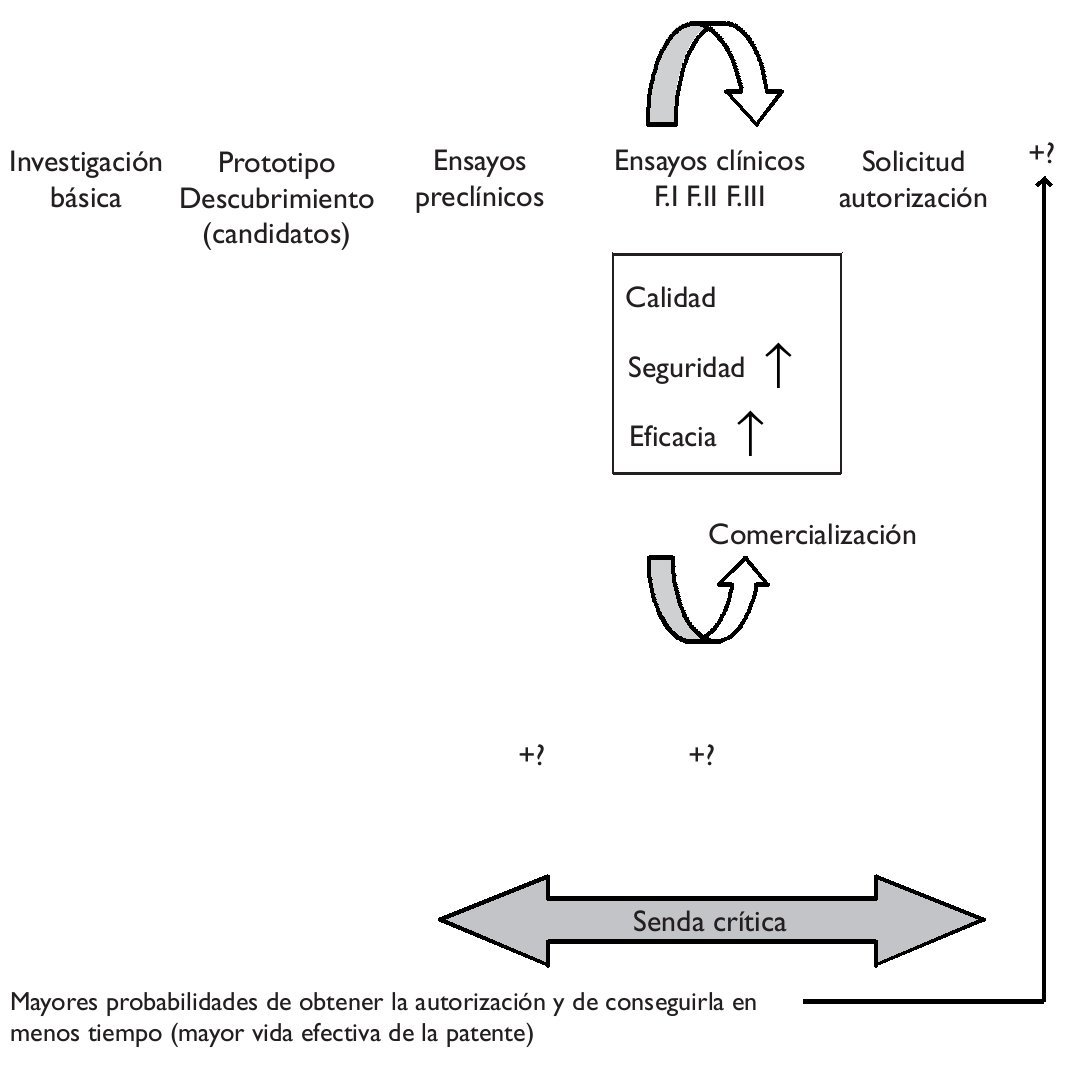
Figura 1."Senda crítica" del desarrollo de un nuevo medicamento e impacto potencial de la farmacogenómica.
En la literatura predomina una visión optimista en relación con la aplicación de la farmacogenómica al proceso de desarrollo de un nuevo medicamento. Se sostiene, por ejemplo, que permite eliminar mucho antes productos no seguros y que la selección de pacientes (con apoyo en biomarcadores/pruebas genéticas) permite aumentar la efectividad y/o disminuir posibles efectos adversos, es decir, aumentar la relación beneficio a riesgo, de modo que es más fácil demostrar el cumplimiento de los requisitos de la eficacia y la seguridad (fig. 1). Se menciona frecuentemente la ventaja que la delimitación de una población diana representa en términos del menor tamaño de los ensayos clínicos, permitiendo reducir costes y grados de incertidumbre. En definitiva, cabría esperar una mayor probabilidad no sólo de conseguir la autorización de una innovación, sino también de obtenerla en menos tiempo, lo que aumentaría la vida efectiva de la patente (el tiempo que le resta, descontado el empleado en conseguir la autorización). No obstante, algunos autores apuntan que si la prevalencia genética es demasiado baja, enrolar en los ensayos clínicos sólo a quienes tienen el marcador podría absorber más tiempo y, en definitiva, acarrear mayores costes2. Del mismo modo, se mencionan costes adicionales en la etapa de los ensayos preclínicos, asociados a la identificación y posterior validación de los biomarcadores3.
Es fundamental hacer hincapié en que la aplicación de la farmacogenómica al proceso de desarrollo de un nuevo fármaco incorpora un elemento nuevo que lo torna aún más complejo; para que la información genómica sea útil el nuevo medicamento precisará de una prueba complementaria. En definitiva, es preciso hacer referencia a la relación de complementariedad medicamento/prueba. Existen tres posibilidades en cuanto a la disponibilidad de la prueba en el tiempo:
1. Disponibilidad previa del nuevo fármaco y posterior de la prueba: la selección de pacientes resultante de la realización de la prueba genética implica una segmentación del mercado (pacientes excluidos por falta de eficacia y/o elevados riesgos de efectos adversos) que amenaza el modelo blockbuster. En este entorno cobran especial relevancia tanto la obtención o adquisición de los derechos de patente de las pruebas, como la ausencia de interés en su explotación por parte del laboratorio farmacéutico, salvo que exista la posibilidad de obtener ventajas competitivas frente a tratamientos alternativos.
2. Disponibilidad previa de la prueba y posterior del nuevo fármaco: ésta puede ser una vía de avance para la obtención de innovaciones (nuevas dianas),así como para el rescate de medicamentos fallidos (por problemas de seguridad y/o eficacia en su día, cuando la indicación había sido planteada de modo menos selectivo).Conviene señalar que esta última posibilidad puede quedar velada por el agotamiento de los derechos de patente de muchos de los medicamentos susceptibles de rescate.
3. Desarrollo simultáneo del nuevo fármaco y de la prueba: la integración del marcador en el ensayo clínico para demostrar la eficacia y/o seguridad del medicamento aporta evidencia sobre la utilidad clínica de la prueba (evidencia en no pocas ocasiones ausente o cuestionada por la literatura).Sin embargo, existen contados ejemplos de desarrollo simultáneo de nuevos principios activos y sus correspondientes pruebas genéticas: trastuzumab, imatinib y cetuximab4,los tres autorizados tanto en EE.UU. como en la Unión Europea (a través del procedimiento centralizado) entre 1998 y 2004.
Los agentes implicados: entre potencialidades y realidades
La farmacogenómica implica numerosos agentes, cuyos intereses no siempre coinciden. Así, el descubrimiento del marcador/prueba y, en su caso, la manufactura del soporte correspondiente, pueden ir asociados tanto a laboratorios farmacéuticos como a empresas de biotecnología o de técnicas de diagnóstico. El agotamiento de los derechos de la patente de medicamentos blockbuster y las dificultades cada vez mayores en su obtención, pueden conducir a la industria tradicional a seguir estrategias (complementarias) apoyadas en la farmacogenómica, a través de operaciones de absorción, alianzas, licencias, adquisición de derechos de patente e incluso procesos de aprendizaje y desarrollo internos. Desde la perspectiva de los laboratorios farmacéuticos la Medicina personalizada implica segmentación del mercado, debido a la exclusión de parte de la población y/o ajuste de dosis y duración del tratamiento. La indicación queda mucho más acotada, de modo que cabe prever que se definan mercados de menor tamaño. Si bien éste es un factor que apunta hacia una menor rentabilidad de la inversión en I+D, el impacto neto depende de muchos otros factores, como la posible consecución de menores costes en el desarrollo del producto; una autorización más rápida del nuevo medicamento, de modo que se disponga de mayor tiempo para explotar la patente; una diferenciación técnica del producto más allá de la marca (algunos autores aluden al valor que el marcador puede añadir al medicamento) y el precio.
El papel del regulador es clave en este contexto, en la medida en la que la Medicina personalizada aconseja adoptar una visión integral de actividades hasta ahora separadas —obtención y comercialización de nuevos medicamentos y de biomarcadores/ pruebas— e históricamente reguladas de forma independiente. Es preciso destacar que la regulación del soporte de las pruebas, tanto en EE.UU. como en la Unión Europea es muy laxa, y que la validación de éstas es una cuestión previa ineludible. A modo de ejemplo, desde la perspectiva de un potencial tercer pagador (sea una compañía privada de seguros, sea el sector público), cabe preguntarse si tiene sentido financiar el medicamento y no la prueba si ésta está validada y permite obtener mejores resultados sobre la salud.
Por último, conviene mencionar las expectativas de los potenciales usuarios y de los clínicos. Las expectativas de la población no siempre se ven satisfechas en la práctica clínica o se cumplen, desde su perspectiva, con retardo. Quizás más en EE.UU. que en Europa el marketing de diversas pruebas por Internet permite prever un fenómeno de oferta inducida. Por su parte, dado que la Medicina personalizada puede afectar sensiblemente sus decisiones, los clínicos precisan apoyo tanto del regulador como del potencial financiador, por ejemplo en términos de validación de las pruebas, formación, diseño de guías, algoritmos, etc. Cabe destacar en este sentido que de la literatura se desprende un elevado grado de desconfianza de los profesionales acerca de la validez de las pruebas genéticas frente a los métodos de seguimiento tradicionales. Surgen pues cuestiones éticas y posibles tensiones con las expectativas de los pacientes y preguntas tales como "¿qué puedo hacer?" (conocimiento científico/fuente de financiación);"¿qué hago?";"¿cómo lo hago?";y "¿qué decisión tomo?" (nótese que no todas las pruebas tienen una interpretación fácil y/o directa).
En definitiva, si bien existen factores asociados a la farmacogenómica que probablemente permitan acortar en algunos casos "la senda crítica", existen otros factores que posiblemente abran un tramo nuevo, sobre todo debido a las particularidades en el uso clínico. Con todo, el modelo sanitario predominante en cada país será, en la práctica, un elemento determinante en la disponibilidad y accesibilidad a productos obtenidos con apoyo en la farmacogenómica. En particular, en un marco de precios administrados como es el español, son muy pertinentes los estudios de evaluación económica, con vistas a apoyar decisiones sobre si procede o no su financiación con fondos públicos y, en caso afirmativo, a qué precio. Se trata de la "cuarta valla" que añadiría a la demostración de la calidad, seguridad y eficacia, la aportación de una comparación de costes e impacto sobre la salud frente a otras alternativas para demostrar una buena relación coste-efectividad. Desde la perspectiva de la industria sería una oportunidad de demostrar el valor del producto en términos de eficacia relativa, tema no baladí si se tiene en cuenta que, tanto en EE.UU. como en la Unión Europea sólo se exige la demostración de la eficacia absoluta (frente a placebo) para conseguir la autorización de un nuevo medicamento. En resumen, en un sistema predominantemente público como es el español, las autoridades tienen responsabilidades de calado en dos ámbitos: validación de las pruebas genómicas y análisis del impacto relativo de terapias apoyadas en las mismas (sobre la salud y sobre los recursos),con vistas a su posible financiación con fondos públicos.
Evaluación económica de intervenciones apoyadas en la Medicina personalizada
Del mismo modo que los estudios de evaluación económica de intervenciones apoyadas en la farmacogenómica son muy pertinentes, su realización puede resultar particularmente compleja debido, entre otras razones, a problemas de incertidumbre y de disponibilidad de datos que, de todos modos, no eximen a las autoridades públicas de la responsabilidad de tomar decisiones. Así,aun disponiendo de una prueba muy fiable para detectar variantes genéticas, una baja penetración del genotipo tiene gran trascendencia en este ámbito, ya que los falsos positivos (o error tipo II) pueden conducir a tratar o a privar de un tratamiento innecesariamente, debido a que la enfermedad o los efectos adversos, finalmente, no se presentan. En la tabla 1 se recogen algunos factores clave que permiten prever (ceteris paribus; si todo lo demás permanece constante) una buena relación coste-efectividad o que, al menos, sugieren el interés de realizar un estudio de evaluación económica en este ámbito.

Con independencia de que sea preciso hacer un análisis caso por caso, hasta el momento existen algunas áreas que parecen especialmente adecuadas para la realización de estudios de estas características. De hecho, una parte nada despreciable está siendo aplicada a enfermedades agudas, sobremanera las relacionadas con la Oncología, área en la que la genómica está permitiendo hacer una subtipificación genética de las enfermedades y en la que se cumplen, entre otras, condiciones como la gravedad de la enfermedad, tratamientos disponibles generalmente muy costosos y con bajo índice terapéutico y elevada variabilidad interpersonal en la respuesta (tabla 1).Respecto a las enfermedades crónicas, el atractivo de la aplicación de este tipo de estudios se refiere a la evitación de muchos años de terapia innecesaria o inadecuada, pero la influencia de numerosos polimorfismos y factores de riesgo inspiran menor optimismo. En resumen, de momento, cabe esperar menor potencialidad en relación con las enfermedades que prácticamente toda la población es susceptible de padecer. No obstante, están surgiendo expectativas positivas en áreas como la Neurología y la Psiquiatría. Así, el hecho de que el seguimiento de la respuesta a los tratamientos actualmente aplicados sea difícil y consuma mucho tiempo conduce a considerar buenas candidatas enfermedades como el Alzheimer o la depresión8.Del mismo modo, la Psiquiatría en general comienza a ser un terreno prometedor, porque varios estudios han mostrado que se puede predecir la respuesta a antipsicóticos y antidepresivos con apoyo en polimorfismos9.
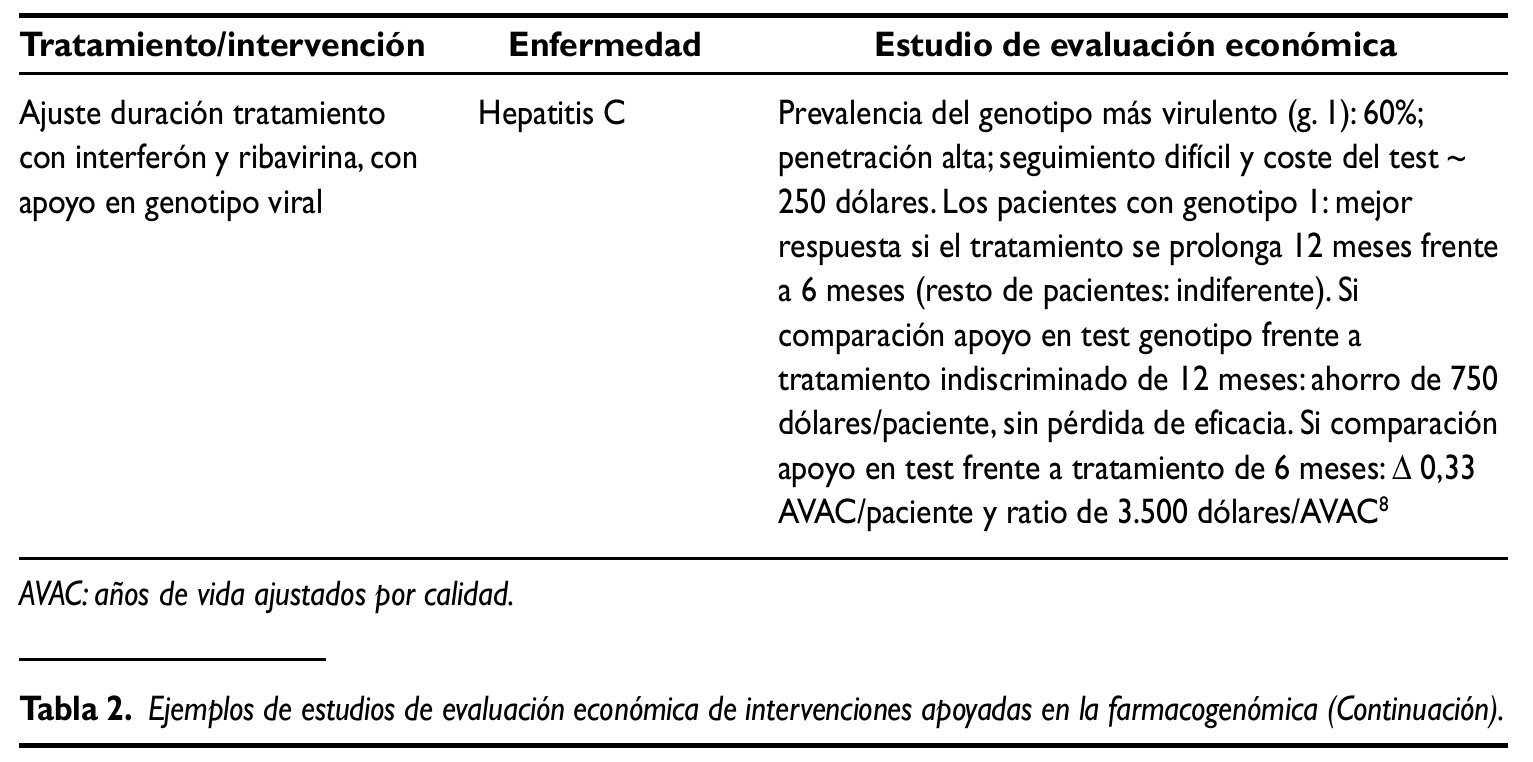
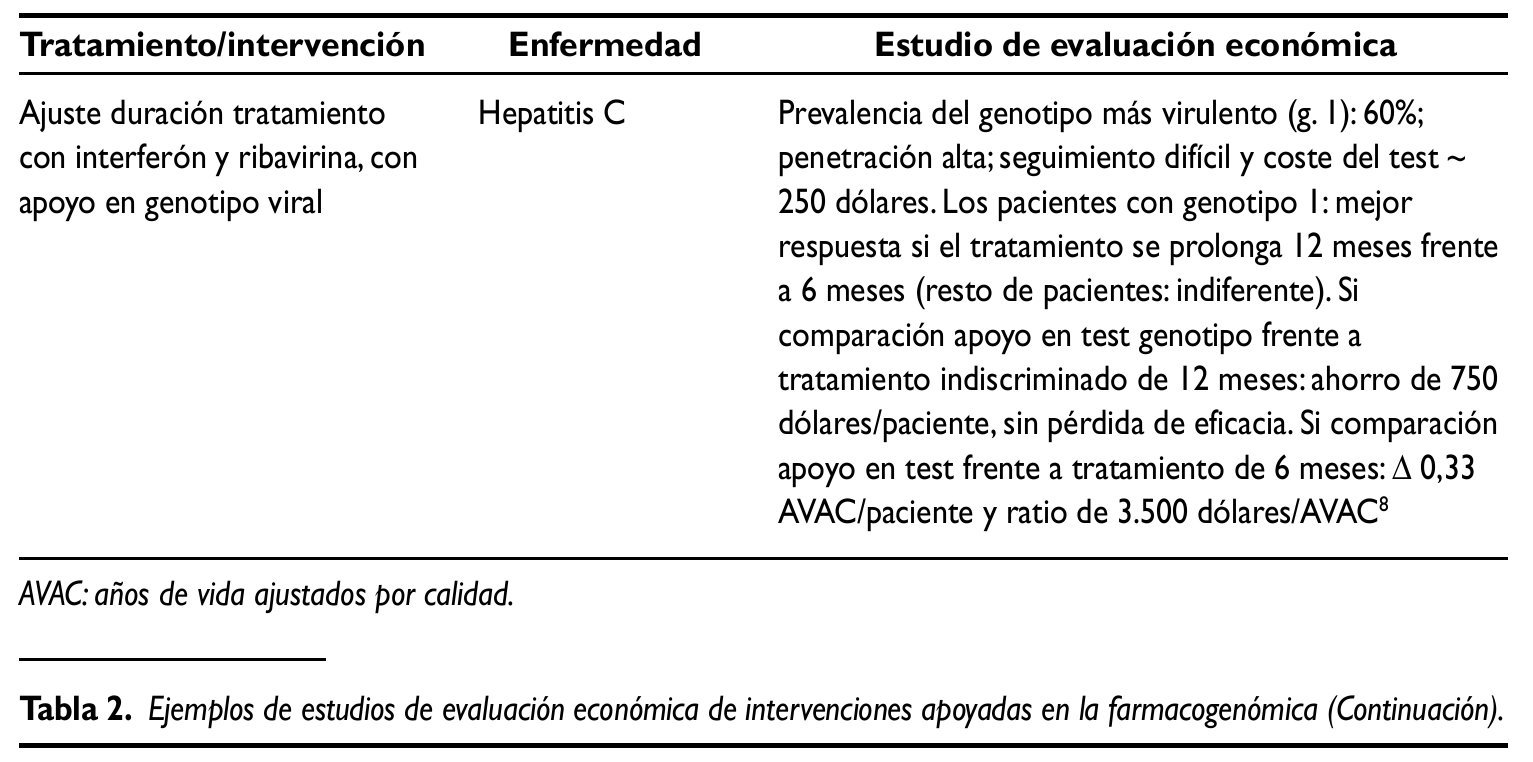
Las mejoras relativas sobre la salud que (en su caso) permiten alcanzar las nuevas tecnologías generalmente van asociadas a mayores costes netos. Dado que sería insostenible financiar todo nuevo tratamiento, en los sistemas sanitarios públicos se puede tomar como referencia la ratio obtenida en el análisis incremental, comparándola con la correspondiente a los tratamientos generalmente aceptados en un marco determinado:30.000 libras/ años de vida ajustados por calidad (AVAC), implícitas en las recomendaciones del NICE; 100.000 dólares/ AVAC en EE.UU.y 30.000 euros/AVAC en España10.Se trata de umbrales considerados en una sociedad y en un momento determinados que, complementados con otras dimensiones, contribuyen a legitimar un proceso de priorización con recursos públicos. A título ilustrativo, en la tabla 2 se presentan sintéticamente los resultados de varios estudios de evaluación económica de intervenciones asociadas a la farmacogenómica. Los dos primeros evalúan el uso de trastuzumab como tratamiento adyuvante de cáncer de mama HER2 positivo. Se trata de un principio activo pionero en el ámbito de la Medicina personalizada, autorizado en 1998 en EE.UU. y en 2000 en la Unión Europea. A pesar de la reducida evidencia evaluadora disponible en general, en este caso y, dado su carácter pionero, existen numerosos estudios de evaluación económica. Se seleccionaron los dos recogidos en la tabla 2, en gran medida porque ambos se aplican al marco sanitario público europeo (Inglaterra/Gales e Italia, respectivamente) y son, hasta cierto punto, comparables. El tercer ejemplo ilustra la posible utilidad de las pruebas genéticas en la elección de tratamiento (en este caso, aplicado al sida). El último estudio recogido consiste en ajustar la duración del tratamiento de la hepatitis C con interferón y ribivirina, con apoyo en el genotipo viral.

A modo de conclusión
De lo expuesto previamente cabe concluir que, para explotar apropiadamente las ventajas de la farmacogenómica desde una perspectiva social, conviene profundizar en algunas parcelas. En el ámbito público-privado la complejidad del tema tratado impone límites prácticos que aconsejan articular nuevos modelos de cooperación, por ejemplo a efectos de obtener la información necesaria para validar un marcador4, de donde se desprende la idoneidad de los consorcios. En el ámbito estrictamente público la Medicina personalizada conduce incluso a replantear viejos problemas: un dato preocupante se refiere a la efectividad de los tratamientos que están siendo aplicados, ya que sólo el 50% de los pacientes responde positivamente a los mismos14,15.Otra vieja cuestión se relaciona con la susceptibilidad que tiene la información genética per se de ser patentada y su impacto sobre la innovación. Por último, tres cuestiones clave en relación con la nueva dimensión que incorpora la farmacogenómica se refieren a la validación de las pruebas, la regulación del soporte de las mismas, con la posibilidad de revisión paralela del medicamento y de la prueba (ya contemplada en EE.UU.) y la posible traslación al terreno legislativo de la evaluación económica, a efectos de guiar la toma de decisiones en materia de financiación de innovaciones con fondos públicos y, en su caso, fijación de precios. En este sentido, cabe diferenciar entre sistemas sanitarios públicos con "legislación blanda";es decir, mención no coercitiva y ausencia de aplicación sistemática y transparente de la evaluación económica (España) y sistemas con "legislación dura" (Ontario y Australia), sin olvidar que, en gran parte del Reino Unido (donde los precios no son objeto de regulación),cabría hablar de un modelo intermedio que, sin ser coercitivo, incorpora incentivos a su aplicación, en la medida en la que las nuevas tecnologías recomendadas por el NICE tienen garantizada la financiación con fondos públicos.