


La hormona luteinizante (LH) y la gonadotropina coriónica humana (hCG) puede inducir esteroidogénesis, hiperplasia y tumorigénesis adrenal a través del estímulo sobre el receptor constitutivo de la LH (R-LHCG) en la corteza adrenal. Los mecanismos fisiopatológicos del síndrome de Cushing adrenal dependiente de LH (SCa-LH) no se han establecido plenamente, pero es reconocida la relación ontogénica adrenal-gonadal con mutua participación de diversos genes, factores de transcripción y enzimas esteroidogénicas como posible causa. El SCa-LH fue descrito en mujeres durante la gestación por el estímulo de hCG y en la posmenopausia ante el aumento de LH, así como en hurones luego de la gonadectomía quirúrgica.
Luteinising hormone (LH) and human chorionic gonadotropin (hCG) can induce steroidogenesis, hyperplasia, and adrenal tumorigenesis through the stimulus of the constitutive LH receptor (R-LHCG) within the adrenal cortex. The pathophysiological mechanisms of luteinising hormone-dependent Cushing's syndrome are not completely understood, but the ontogenic relationship between the adrenal cortex and the gonads, with mutual participation of different genes, transcription factors and steroidogenic enzymes cited as a possible cause, is well-recognised. SCa-LH has been described in pregnant women, as a result of hCG stimulus, and in post-menopausal women, due to increased levels of LH, as well as in ferrets after gonadectomy.
Con el descubrimiento de los anticuerpos monoclonales, el desarrollo de la inmunohistoquímica y el avance de la biología molecular se ha logrado aclarar varios mecanismos patológicos con referencia al síndrome de Cushing adrenal (SCa), anteriormente denominado «ACTH-independiente» (sigla en inglés de adrenocorticotropic hormone o adrenocorticotropina). Se han estudiado diferentes etiologías del SCa, una de las cuales, la presencia de receptores ectópicos o eutópicos de diferentes hormonas con sobreexpresión en la corteza adrenal, ha sido motivo de investigaciones recientes. El hallazgo de estos receptores y su asociación con el SCa ha obligado a ampliar la exploración diagnóstica y a considerar nuevas manifestaciones clínicas y subclínicas de presentación. El objetivo de esta monografía es caracterizar el SCa-LH, resaltando la íntima e importante relación ontogénica adrenal-gonadal.
Síndrome de CushingEl síndrome de Cushing (SC) endógeno hace referencia a la constelación de anomalías clínicas y bioquímicas que resultan de la exposición crónica al exceso de cortisol producido por la corteza adrenal. Entre el 80 y el 85% del SC es causado por un adenoma hipofisario productor de ACTH, que se denomina «enfermedad de Cushing» (EC) Con menos frecuencia puede ocurrir por un tumor extrahipofisario (síndrome de ACTH ectópico) o, muy raramente, por un tumor secretor de hormona liberadora de corticotropina (CRH, por su sigla en inglés). El SC también puede ser adrenal (SCa), entre el 15 y el 20% de los casos, cuando el exceso de secreción de cortisol derivó de un adenoma adrenocortical (AA), carcinoma adrenocortical (CA), hiperplasia adrenocortical macronodular bilateral primaria (BMAH, por su sigla en inglés), enfermedad adrenocortical micronodular no pigmentada (MAD, por su sigla en inglés) y enfermedad adrenocortical micronodular pigmentada primaria (PPNAD, por su sigla en inglés)1. En el ser humano, el SC es un trastorno raro, con una incidencia estimada de 1,2 a 1,7 nuevos casos por millón de habitantes/año2.
En el perro, el SC se presenta con una alta incidencia, 1 a 2 casos/1.000 perros/año y, al igual que en el humano, cerca del 80 al 85% de los casos son por EC3. Aproximadamente del 71 al 80% de los tumores hipofisarios en esta especie surgen de la parte distal; el resto es originado en la parte intermedia3. No se han informado casos de SC por tumor secretor de CRH y la secreción de ACTH ectópica es muy rara3,4. El SCa en el perro también puede derivar de AA, CA o BMAH3.
Síndrome de Cushing adrenalEl AA y el CA unilateral comprenden el 90% de los casos de SCa1, mientras que el restante 10% lo constituyen los diferentes tipos de hiperplasia nodular adrenocortical5.
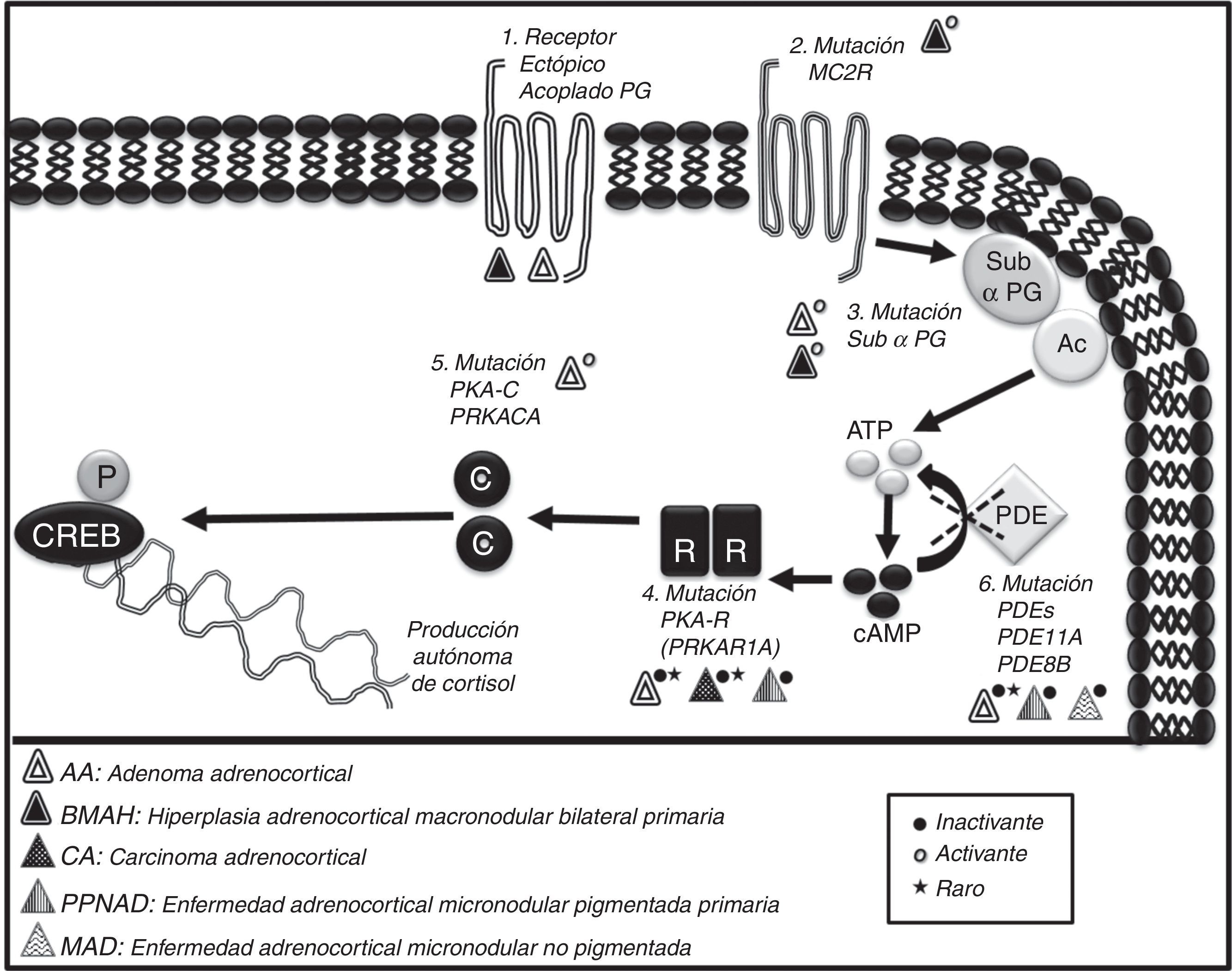
El 55% de los tumores adrenales (TA) corresponde a un AA y en ellos se identificaron mutaciones activantes en los genes PRKACA (subunidad catalítica de la proteína cinasa A)6 y GNAS1 (subunidad α de la proteína G)7, así como mutaciones inactivantes en los genes PRKAR1A (subunidad reguladora de la proteína cinasa A) y PDE8B (fosfodiesterasa)8, y una mutación en la vía de señalización de Wnt/β-catenina (gen CTNNB1) con aumento de la transcripción celular9.
En el CA, las alteraciones moleculares más frecuentes se presentan en la vía de señalización Wnt/β-catenina, mutaciones en el gen supresor de tumores p53 y el gen RB1 (asociado al retinoblastoma); la alteración en la vía del monofosfato cíclico de adenosina (AMPc) es rara10.
Recientemente fue descubierta la producción y regulación de ACTH intraadrenal en la hiperplasia adrenocortical macronodular independiente de ACTH (AIMAH, por su sigla en inglés), y con este hallazgo la AIMAH fue redefinida como «BMAH»11,12. En su etiología se identificó una mutación germinal inactivante en el gen supresor de tumores ARMC513 y mutaciones activantes en MC2R (receptor de melanocortina)14 y GNAS1, la última de las cuales está asociada con el síndrome de McCune-Albright15.
Se describen 2 formas de hiperplasia micronodular, la MAD y la PPNAD16. Los casos de MAD se asocian con mutaciones inactivantes en genes de la fosfodiesterasa PDE (PDE11A, PDE8B)16 (fig. 1). La variable PPNAD ha sido atribuida a una mutación germinal inactivante en el gen PRKAR1A, asociada con el 60% de los pacientes con el complejo de Carney (CNC en inglés)17. La PPNAD no asociada a CNC o aislada ha sido también vinculada a mutaciones inactivantes en PRKAR1A, así como a una alteración en los genes de la fosfodiesterasa PDE11A y PDE8B16.

Características moleculares del síndrome de Cushing adrenal.Adaptado de Duan et al.5.
En el perro, un tercio de los TA presenta una mutación activante de GNAS13.
En el caso de AA, BMAH y, raramente, CA, fueron identificados receptores de membrana acoplados a proteína G (GPCR, en inglés) aberrantes (ectópicos o eutópicos con sobreexpresión)18.
Síndrome de Cushing por receptores aberrantes en corteza adrenalLa ACTH normalmente es la única hormona que estimula la adenilato-ciclasa a través de un GPCR y, por consiguiente, la esteroidogénesis adrenal. Sin embargo, otras hormonas con unión a GPCR aberrantes en TA y BMAH también lo estimulan y generan SCa18. En ratas, se encontró en la corteza adrenal la presencia de múltiples receptores de adrenalina, noradrenalina, hormona estimulante de la tiroides, LH y hormona foliculoestimulante19. Desde entonces, se han descrito en la corteza adrenal receptores ectópicos β-adrenérgicos (B-AR) en CA de ratas y humanos con BMAH18,20, al péptido inhibidor gástrico (GIP-R) en AA18,21 y BMAH3,18,22, a la vasopresina 2 (AVPR2) y vasopresina 3 (AVPR3) en BMAH18 y 5 hidroxitriptamina (5-HT) 5-HT718. También fue descrita la sobre expresión de receptores eutópicos para LH (R-LHCG) en BMAH18,23, vasopresina 1 (AVPR1)18, 5-HT (5-HT4)18 y glucagón en BMAH24.
En el perro, R-LHCG y AVPR1 se expresaron en glándulas adrenales normales (GAn) considerándose eutópicos, mientras que GIP-R y AVPR2 fueron considerados ectópicos por expresarse solo en TA secretores de cortisol3.
Síndrome de Cushing adrenal dependiente de hormona luteinizanteEl receptor R-LHCG se denomina así debido a que puede unirse con alta afinidad tanto a la LH como a la hCG, y aunque clásicamente se expresa en las gónadas25, también se lo ha identificado constitutivamente y con diferentes grados de expresión en la corteza adrenal de GAn del humano, hurón, perro y roedores, aunque aún no llega a comprenderse por completo su función25. Sin embargo, ha sido encontrado en AA, CA y BMAH, sugiriéndose su protagonismo en el desarrollo del SCa18.
En el ser humano, el R-LHCG ha sido vinculado al SCa-LH en mujeres con posmenopausia y durante la gestación26, mientras que en el hurón se ha relacionado con la gonadectomía quirúrgica27. En cualquiera de los casos mencionados, la producción excesiva de cortisol sería la consecuencia del estímulo crónico sobre el R-LHCG por parte de la LH o la hCG.
Los pacientes con SCa-LH se presentan con mayor frecuencia en el contexto del SC subclínico, cuyos signos predominantes son hipertensión, pérdida de masa muscular proximal, depresión y ganancia de peso22. En la bioquímica, hay insuficiente supresión del cortisol plasmático en el test de Nugent, con ACTH plasmática inhibida o baja22. La confirmación del diagnóstico se realiza al obtener aumento del cortisol plasmático luego del estímulo con LH, hCG u hormona liberadora de gonadotropina (GnRH)22. Finalmente, la determinación de BMAH, AA o CA se realiza mediante tomografía computarizada5.
SC dependiente de LH en el hurón: la prevalencia del SCa-LH en los Estados Unidos fue mayor del 70% en el año 200328. En esta especie se describió la presencia de hiperplasia corticoadrenal en el 56%, AA en el 16% y CA en el 26%, en todos los casos con expresión de R-LHCG28. La etiología del SCa-LH ha sido atribuida a factores genéticos, ovariohisterectomía y fotoperiodo artificialmente prolongado. Tanto la gonadectomía como el aumento en las horas de luz (disminución de melatonina) conllevan el aumento de GnRH hipotalámico con aumento de LH27. El diagnóstico del SCa se establece con el aumento de la relación cortisol/creatinina urinaria (UCCR en inglés), valores de ACTH bajo o suprimido y la presencia de hiperplasia o neoplasia de las glándulas adrenales por ecografía. El diagnóstico de SCa-LH se realiza al obtener aumento de cortisol plasmático luego del estímulo con hCG. Los implantes de depósito con agonistas de GnRH (leuprolide o deslorelina) son utilizados como tratamiento de esta patología, ya que desensibilizan el receptor de GnRH, con posterior disminución de gonadotropinas28.
Estrecha relación ontogénica adrenal-gonadal: el primordio adrenal fetal (células esteroidogénicas de la corteza adrenal) y el primordio gonadal sexual (células esteroidogénicas de las gónadas) derivan de un precursor común mesodérmico denominado «primordio adrenogonadal» (PAG). En dichos primordios fueron rastreados los factores de transcripción del factor esteroidogénico tipo 1 (SF-1) y DAX-1, los cuales fueron vinculados como responsables de la formación, la diferenciación y el mantenimiento del desarrollo adrenal y gonadal, además del control de la esteroidogénesis y la regulación de los genes implicados en la cascada determinante del sexo29. Las alteraciones en dichos factores han llevado a la insuficiencia adrenal y seudohermafroditismo (Phf) masculino en ratones por deficiencia de SF-130 y a hipoplasia suprarrenal congénita ligada al X e hipogonadismo hipogonadotrófico en ambos sexos por alteración en DAX-130.
Otra evidencia de la estrecha relación genética adrenal-gonadal se presenta cuando se dan alteraciones en la regulación de la actividad enzimática (esteroidogénesis) en enfermedades como hiperplasia suprarrenal lipoide y Phf en el feto masculino (gen StAR, enzima colesterol desmolasa 20-22)31, hiperplasia suprarrenal congénita (HSC) y Phf masculino (gen 3BHSD tipo II, déficit 3BHSD2)32, y Phf e HSC (gen CYP17, déficit de 17 α hidroxilasa/17,20 desmolasa)33. Otros genes, como CYP11B1, CYP21A y MC2R, expresados específicamente en la corteza adrenal de adultos, también son expresados en el testículo fetal29. Las aromatasas específicas de gónadas adultas CYP19A1A y CYP17 también son expresadas en la glándula suprarrenal fetal34.
Neoplasia adrenocortical inducida por gonadectomía: la expresión de SF-1 y GATA-6, y la baja expresión de DAX-1 sobre las células madre subcapsulares de la corteza adrenal adulta generan la diferenciación del fenotipo celular adrenal35. La estimulación crónica de LH sobre el R-LHCG conduce al aumento de GATA-4 (factor de transcripción normalmente ausente en adrenales y presente en gónadas) y, junto con la expresión de SF-1, lleva a la metaplasia de las células de la corteza adrenal, transformándolas en un tejido similar al estroma gonadal, denominado «metaplasia tecal»36,37. Además, GATA-4 estimula la regulación positiva de genes expresados en células somáticas de gónadas que codifican para la hormona antimülleriana, proteína reguladora aguda esteroidogénica, inhibina α, inhibina β y 17 hidroxilasa/17-20 liasa. Esta regulación positiva de genes permite la respuesta adrenal al aumento de LH. GATA-4 también aumenta la expresión del R-LHCG, haciendo a este tejido más sensible a la acción de la LH37. En TA, se ha encontrado la expresión de GATA-4 y existe evidencia de que su ausencia atenúa la aparición de TA en ratones38.
SCa dependiente de LH sensible a la serotonina: en individuos sanos, la serotonina intraadrenal estimula la secreción de aldosterona (expresión de receptor 5-HT4 en zona glomerular) pero no la de cortisol (baja expresión 5-HT4 en zona fascicular). Es probable que la LH induzca la liberación de serotonina intraadrenal y esta, por sí misma, la producción de cortisol. Una explicación de este fenómeno podría ser que se produzca un aumento en la expresión del receptor 5-HT4 en las células productoras de cortisol por LH, o que exista un acoplamiento funcional alterado entre el receptor 5-HT4 y la secreción de cortisol. La serotonina también puede influir en la producción de cortisol indirectamente, vía inducción de la liberación intraadrenal de interleucina-639.
ConclusionesLa presencia constitutiva del R-LHCG en la corteza adrenal y el origen ontogénico común, que involucra factores de transcripción, genéticos y enzimáticos de la glándula adrenal y las gónadas, constituyen una explicación plausible de por qué la LH y la hCG pueden llegar a inducir la esteroidogénesis y tumorigénesis adrenal.
Es claro que solamente la presencia del R-LHCG no es suficiente para el desarrollo del SCa-LH: se requiere un estímulo crónico de LH sobre este, como ocurre en la menopausia. Sin embargo, no todas las mujeres posmenopáusicas desarrollan SCa-LH, lo que indica la existencia de otros factores implicados en su desarrollo, todavía no identificados.
En el caso de la hCG durante la gestación, se sabe que se une al R-LHCG, pero el mecanismo exacto de desarrollo del SCa-LH es más desconocido que en el caso de la LH.
En el hurón, el aumento de LH posgonadectomía, junto con la expresión constitutiva de R-LHCG, genera SCa-LH, pero en esta especie se presenta con una alta prevalencia, por lo que se debe considerar la gonadectomía a edades tempranas como un fuerte factor desencadenante; incluso se podría considerar que este hecho produzca una exposición crónica prematura de LH sobre su receptor en la corteza adrenal o que ciertos factores de transcripción expresados solamente en la edad fetal sean prontamente activados por el estímulo temprano de LH.
Por otro lado, es probable que exista una interacción entre LH, serotonina y sus receptores en la regulación de la producción del cortisol; sin embargo, el mecanismo exacto es desconocido actualmente.
Finalmente, se recomienda en el perro con SCa e hipogonadismo hipergonadotrófico posgonadectomía quirúrgica la implementación de la evaluación diagnóstica de SCa-LH mediante pruebas de estímulo con hCG, LH o GnRH.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores no poseen conflictos de interés.
A mis compañeros de la Unidad de Endocrinología en el Hospital Escuela de la Facultad de Ciencias Veterinarias de la Universidad de Buenos Aires, por todas sus enseñanzas. A mi familia en Argentina (María Elisa Caneda y Vincent) y en Tunja, Boyacá, Colombia, por su apoyo incondicional.