



La listeriosis humana es una enfermedad infecciosa causada por Listeria monocytogenes. La forma invasiva presenta una alta tasa de hospitalizaciones y mortalidad. El principal modo de transmisión es a través de alimentos listos para el consumo contaminados, como lácteos, vegetales y productos cárnicos. El conocimiento de la diversidad y dinámica poblacional de aislamientos recuperados de humanos y matrices alimentarias resulta fundamental para detectar conglomerados e identificar sitios comunes de infección. El objetivo de este estudio fue caracterizar desde el punto de vista molecular aislamientos de L. monocytogenes en Argentina. Se secuenciaron 63 aislamientos en total, 35 de origen humano y 28 aislados de alimentos, recolectados entre 2018 y 2023. El estudio genómico dividió los aislamientos en dos linajes, cuatro serogrupos, 17 secuenciotipos y 15 complejos clonales (CC). El clon hipervirulento CC1 (linaje I; serogrupo IVb) predominó en muestras de humanos y de alimentos. El análisis filogenómico basado en la detección de polimorfismos de nucleótido único (SNP) detectó elevada y posible relación epidemiológica entre aislamientos de humanos y de alimentos. Ello sugiere la existencia de cadenas de transmisión en nuestro medio. Estos hallazgos destacan la necesidad de fortalecer la vigilancia genómica de L. monocytogenes en Argentina. Conocer la distribución geográfica y las características de los clones predominantes y emergentes recuperados de humanos y de matrices alimentarias permitirá elaborar planes de acción y políticas de salud pública mejor dirigidas para el control y la prevención de la listeriosis.
Human listeriosis is an infectious disease caused by Listeria monocytogenes. The invasive form of this disease leads to a high rate of hospitalizations and fatality. The main mode of transmission is through contaminated ready-to-eat foods such as dairy, vegetables and meat products. The knowledge of the diversity and population dynamics of isolates collected from human and food sources is essential for the detection of clusters and the identification of common sites of infection. The aim of this study was the molecular characterization of L. monocytogenes isolates in Argentina. We sequenced a total of 63 isolates, 35 from human and 28 from food sources, collected between 2018 and 2023. Our genomic study divided the isolates into two lineages, four serogroups, 17 sequence types and 15 clonal complexes (CCs). The hypervirulent clone CC1 (lineage I; serogroup IVb) predominated in human and food samples. The phylogenomic analysis showed a high and possible epidemiological relationship between isolates from human and/or food sources, suggesting the presence of transmission chains in our country. These findings highlight the need to strengthen genomic surveillance of L. monocytogenes in Argentina. The identification of geographic distribution and characteristics of predominant and emerging clones from human and food sources might help to focus action plans and public health policies better directed at the control and prevention of listeriosis.
Listeria monocytogenes es el agente causal de la listeriosis humana, una de las enfermedades de transmisión alimentaria más graves que afectan a los humanos12. L. monocytogenes es un patógeno intracelular facultativo de humanos y animales y está ampliamente distribuido en el medioambiente12. La contaminación de los alimentos por esta bacteria puede originarse en cualquier momento de la cadena de producción hasta el consumo. Su capacidad para sobrevivir en superficies de contacto y condiciones de estrés (ej., secado, refrigeración y acidez) facilita su persistencia y diseminación3,13.
La listeriosis gastrointestinal no invasiva es la presentación clínica más frecuente21. Sin embargo, la forma más grave de la infección es la listeriosis invasiva, que puede presentarse como encefalitis, meningitis y sepsis, con una mortalidad estimada del 20 al 30%4. Afecta mayormente a individuos con debilitamiento del sistema inmune, como mujeres embarazadas, neonatos, pacientes de edad avanzada e inmunocomprometidos4. Su capacidad de resistencia y adaptación al medioambiente y su elevada letalidad convierten a L. monocytogenes en una de las mayores problemáticas de la industria de alimentos y la salud pública.
La vigilancia epidemiológica de los casos humanos y las matrices alimentarias resulta fundamental para detectar conglomerados e identificar sitios comunes de infección25. Además, proporciona información relevante para la adopción temprana de medidas preventivas de control y protección. L. monocytogenes es una especie heterogénea, la cual se agrupa en cuatro linajes evolutivos y 14 diferentes serotipos12,22. Además, mediante tipificación por secuenciación de múltiples locus (MLST, por su sigla en inglés), pueden ser divididos en múltiples complejos clonales (CC) y secuenciotipos (ST)23. Los complejos clonales CC1, CC2, CC4 y CC6, del linaje I, se consideran hipervirulentos y se asocian con un mayor número de infecciones graves en humanos, mientras que los complejos clonales CC9 y CC121, del linaje II, son considerados hipovirulentos e identificados, principalmente, en alimentos5,16,17. Los aislamientos del CC3, CC5, CC7 y CC155 son considerados intermedios y, en estudios anteriores, fueron encontrados tanto en alimentos como en muestras clínicas5,16,17. En Argentina, se dispone de pocos datos sobre la prevalencia e incidencia de la listeriosis y las características de los aislamientos provenientes de distintas fuentes.
El objetivo de este estudio fue realizar la caracterización molecular de aislamientos de L. monocytogenes obtenidos de humanos y alimentos en el periodo 2018-2023 en Argentina.
Materiales y métodosSe estudiaron 63 aislamientos (humanos, n=35; alimentos, n=28) consecutivos derivados al Laboratorio Nacional de Referencia desde distintos centros asistenciales del país, públicos y privados, en el periodo enero/2018 a octubre/2023. Los aislamientos fueron cultivados en agar sangre de carnero, con incubación a 36±1°C en atmósfera enriquecida con 5% de CO2 durante 20-24 h. La identificación a nivel de especie se realizó por espectrometría de masas MALDI-TOF MS (Microflex®, Bruker Daltonics), según las recomendaciones del fabricante, utilizando la base de datos Biotyper 3.1 (Bruker Daltonics) para la interpretación de los resultados. Los aislamientos fueron conservados a –80°C en caldo tripticasa soja con 20% de glicerol.
Secuenciación de genoma completoPara caracterizar molecularmente a los aislamientos y estudiar su relación clonal, se realizó la secuenciación de genoma completo (SGC). La extracción del ADN genómico se realizó utilizando el sistema comercial QIAamp DNA Mini Kit (Qiagen, Hilden, Alemania). El ADN extraído se cuantificó mediante el método fluorométrico Qubit® 2.0 Fluorometer (Thermo-Fisher Scientific, Waltham, MA, EE. UU.) y las muestras de ADN se conservaron a –20°C hasta su procesamiento.
Para la SGC, se utilizó el sistema comercial Nextera XT DNA LibraryPrep y la plataforma Illumina MiSeq (Illumina, San Diego, CA, EE. UU.). La calidad de las secuencias obtenidas se evaluó mediante el programa FastQC 0.11.9 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) y las contaminaciones se identificaron con la herramienta Kraken2 v2.0.629. El ensamblado de novo de los fragmentos de lectura se realizó mediante el software Unicycler v.0.4.8, el cual se basa en SPAdes graphs v.3.13.02. Para el control de calidad de los ensamblados, se utilizó la herramienta Quast v5.0.2, que arrojó un valor medio del número de contigs de 34 y un N50 de 477168 pb8. La presencia de clones circulantes se determinó a partir de los ensamblados de novo mediante el análisis in silico de los genes utilizados por el método de MLST. La base de datos BIGSdb-Lm (https://bigsdb.pasteur.fr/listeria/) permitió determinar el secuenciotipo, el complejo clonal y el linaje. La identificación de serogrupo se realizó a partir de las secuencias de lectura obtenidas mediante la base de alelos de Doumith et al., disponible en BIGSdb-Lm, y la herramienta SRST2 v0.1.87,9.
Análisis filogenómicoLa relación filogenética de los aislamientos se investigó mediante el análisis de polimorfismos de nucleótido único (SNP) contra genoma de referencia, utilizando la cepa EGD-e, depositada en el Bio-proyecto PRJNA276 (NCBI). La herramienta Snippy v4.4.5 (https://github.com/tseemann/snippy) se utilizó para el mapeo de los fragmentos de lectura de los aislamientos al genoma de referencia y la identificación de SNP. El reconocimiento y remoción de sitios de recombinación se realizó mediante Gubbins v 2.1.06. El alineamiento final, que abarcó 50080 sitios, se utilizó para la construcción de la filogenia molecular utilizando IQ-tree v1.6.1, con un modelo de evolución GTR+I+G y estableciendo la confiabilidad de la topología mediante bootstrap con 10000 réplicas20.
La biblioteca Rhierbaps v1.1.2 en el software R v.3.5.1 se utilizó para determinar la presencia de cadenas de transmisión, estableciendo 2 niveles de discriminación y un número de clústeres de 2026. La diferencia de SNP entre los aislamientos se determinó mediante la herramienta snp-dists v0.6.2. Se adjudicó el tipo de relación epidemiológica sobre la base de dicha diferencia: alta relación epidemiológica (0-9 SNP), posible relación epidemiológica (10-29 SNP) y ocasional relación epidemiológica (30-100 SNP)10.
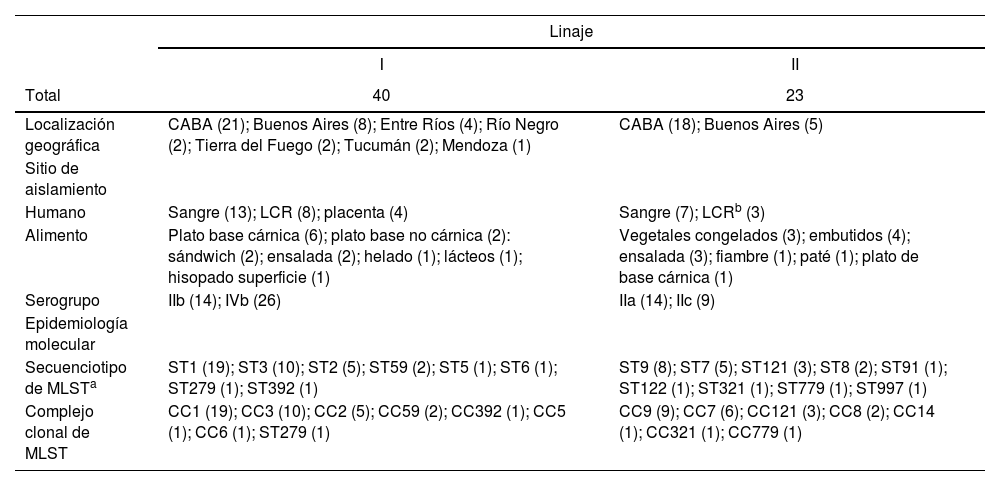
ResultadosEl total de casos humanos fue de origen invasivo y L. monocytogenes se recuperó de hemocultivo (n=20), líquido cefalorraquídeo (n=11) y placenta (n=4). Los aislamientos de origen alimentario se obtuvieron, principalmente, de productos cárnicos listos para el consumo (n=7), ensaladas (n=5), embutidos (n=4) y vegetales congelados (n=3). Los aislamientos provinieron de 7 de las 24 jurisdicciones de Argentina (23 provincias más CABA). La Ciudad Autónoma de Buenos Aires (CABA) (61,9%) y la provincia de Buenos Aires (20,6%) aportaron la mayor parte de los aislamientos; un bajo porcentaje de las muestras provinieron de las provincias de Entre Ríos (6,3%), Tierra del Fuego (3,2%), Río Negro (3,2%), Tucumán (3,2%) y Mendoza (1,6%) (tabla 1).
Características epidemiológicas y moleculares de 63 aislamientos de L. monocytogenes provenientes de muestras de alimentos y humanos en Argentina
| Linaje | ||
|---|---|---|
| I | II | |
| Total | 40 | 23 |
| Localización geográfica | CABA (21); Buenos Aires (8); Entre Ríos (4); Río Negro (2); Tierra del Fuego (2); Tucumán (2); Mendoza (1) | CABA (18); Buenos Aires (5) |
| Sitio de aislamiento | ||
| Humano | Sangre (13); LCR (8); placenta (4) | Sangre (7); LCRb (3) |
| Alimento | Plato base cárnica (6); plato base no cárnica (2): sándwich (2); ensalada (2); helado (1); lácteos (1); hisopado superficie (1) | Vegetales congelados (3); embutidos (4); ensalada (3); fiambre (1); paté (1); plato de base cárnica (1) |
| Serogrupo | IIb (14); IVb (26) | IIa (14); IIc (9) |
| Epidemiología molecular | ||
| Secuenciotipo de MLSTa | ST1 (19); ST3 (10); ST2 (5); ST59 (2); ST5 (1); ST6 (1); ST279 (1); ST392 (1) | ST9 (8); ST7 (5); ST121 (3); ST8 (2); ST91 (1); ST122 (1); ST321 (1); ST779 (1); ST997 (1) |
| Complejo clonal de MLST | CC1 (19); CC3 (10); CC2 (5); CC59 (2); CC392 (1); CC5 (1); CC6 (1); ST279 (1) | CC9 (9); CC7 (6); CC121 (3); CC8 (2); CC14 (1); CC321 (1); CC779 (1) |
En todos los casos, se indica entre paréntesis el número de aislamientos.
A partir del estudio molecular, se determinó que los aislamientos correspondieron a dos linajes predominantes: linaje I (n=40), el mayoritario e identificado en todas las jurisdicciones, y el linaje II (n=23), solo detectado en Buenos Aires y la CABA. Ambos linajes fueron clasificados en cuatro serogrupos. El serogrupo IVb (n=26) fue el mayoritario, seguido por los serogrupos IIb (n=14), IIa (n=14) y IIc (n=9). Los serogrupos IVb y IIb se asociaron al linaje I y los serogrupos IIa y IIc al linaje II. El análisis de MLST reveló 17 ST entre los 63 aislamientos estudiados, de los cuales 9 fueron representados por solo un aislamiento cada uno. El ST1 (n=19) fue el secuenciotipo predominante, seguido por ST3 (n=10), ST9 (n=8), ST2 (n=5) y ST7 (n=5). Además, los ST fueron agrupados en 15 CC, entre los cuales los más comunes fueron el CC1 (n=19), CC3 (n=10), CC9 (n=9), CC7 (n=6) y CC2 (n=5) (tabla 1).
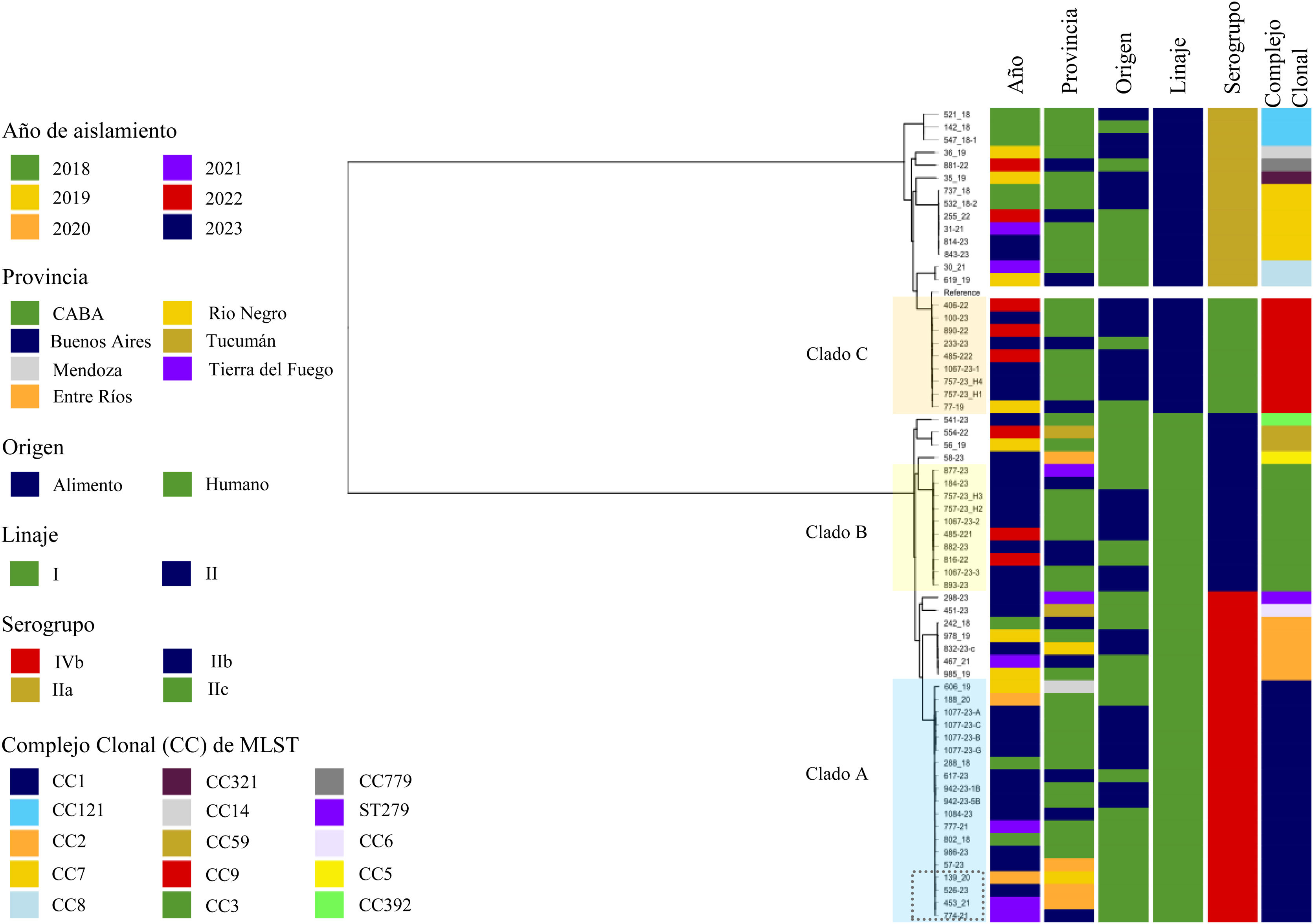
El análisis bayesiano de la filogenia reveló la presencia de 10 clados. Sin embargo, el 61,9% de los aislamientos se agrupó en tres clados, A, B y C, los cuales incluyeron entre 19 y 9 aislamientos (fig. 1).
, el linaje, el serogrupo y el complejo clonal asignado mediante MLST. La caja de línea de puntos (clado A) indica los cuatro aislamientos con alta relación epidemiológica (<10 SNP).")
Árbol filogenómico de 63 aislamientos de L. monocytogenes colectados de muestras de humanos y alimentos en Argentina, 2018-2023. A la derecha del árbol, se indica mediante colores el año y la zona geográfica de aislamiento, el origen (humano/alimento), el linaje, el serogrupo y el complejo clonal asignado mediante MLST. La caja de línea de puntos (clado A) indica los cuatro aislamientos con alta relación epidemiológica (<10 SNP).
El clado A (n=19) se correspondió con el clon hipervirulento CC1, serogrupo IVb y linaje I. Hubo una mayor asociación de este clado con casos humanos (n=12) comparado con alimentos (n=7) y fue detectado en casi todo el período de estudio. Las muestras de alimentos se obtuvieron en la CABA; las de humanos se detectaron en la CABA (n=4), Buenos Aires (n=3), Entre Ríos (n=3), Mendoza (n=1) y Río Negro (n=1). La diferencia media de SNP dentro de este clado fue de 58,7 (rango: 1-102). En este clado, 4 aislamientos recuperados de humanos entre 2020 y 2023, provenientes de Entre Ríos (n=2), Buenos Aires (n=1) y Río Negro (n=1), mostraron una alta relación epidemiológica, con una diferencia de 1 a 9 SNP (fig. 1).
El clado B (n=10) incluyó al CC3, serogrupo IIb y linaje I. Los aislamientos de este clado fueron recolectados entre 2022 y 2023 en la CABA (n=6), Buenos Aires (n=3) y Tierra del Fuego (n=1). Las muestras fueron mayormente de origen alimentario (n=6). Los alimentos implicados fueron productos listos para el consumo (ensaladas, platos de base cárnica y no cárnica y lácteos). La diferencia media de SNP dentro de este clado fue de 40,8 (rango: 11-69). Dos aislamientos de humanos provenientes de Buenos Aires y uno de alimentos, los tres recuperados en la CABA, mostraron una posible relación epidemiológica, con una diferencia de entre 11 y 14 SNP.
El clado C (n=9) estuvo representado por el clon hipovirulento CC9, serogrupo IIc y linaje II. La diferencia media de SNP dentro de este clado fue de 44,7 (rango: 7-64). Las muestras implicadas en el hallazgo de este clado fueron principalmente de alimentos (n=7) y obtenidas en la CABA, mientras que las muestras de humanos (n=2) provinieron de Buenos Aires. El origen de los alimentos fue mayormente productos cárnicos como embutidos (n=4) y fiambre (n=1). Una muestra de humano mostró una posible relación epidemiológica con tres muestras de alimentos, con una diferencia de entre 11 y 14 SNP.
DiscusiónEl concepto de «una salud» se define como una estrategia integrada y unificadora con el objetivo de mejorar la salud de las personas, los animales y los ecosistemas de manera sostenible1,24. Se considera a L. monocytogenes uno de los patógenos bacterianos transmitidos por alimentos y animales infectados más importantes y una especie crítica dentro del marco de una salud12. La capacidad de persistir en distintas condiciones de conservación convierte a este patógeno en una grave amenaza para la seguridad alimentaria13. Por lo tanto, el abordaje de salud pública de este patógeno, dentro del enfoque de una salud, requiere datos sólidos de vigilancia de fuentes humanas, alimentarias, animales y ambientales.
Actualmente, SGC permite un elevado nivel de resolución y se considera la técnica de elección para la vigilancia epidemiológica de L. monocytogenes10,14. Esta permite detectar brotes, evaluar la dinámica poblacional de aislamientos con distintos perfiles de virulencia e identificar reservorios o sitios comunes de infección. En este estudio, el análisis genómico proporcionó una descripción general de los clones de L. monocytogenes circulantes en Argentina y permitió, además, la identificación y asociación de los mismos a distintos grados de virulencia y fuentes en común.
La caracterización molecular mostró una mayor asociación de los aislamientos humanos al CC1 (serogrupo IVb), seguido por el CC3 (serogrupo IIb) y CC7 (serogrupo IIa). En particular, el CC1 es considerado el complejo clonal clínico más prevalente en distintas regiones geográficas y ha sido fuertemente vinculado con el ganado vacuno y los productos lácteos19. La mayor adaptación al huésped, la persistencia y el neurotropismo hacen del CC1 un clon hipervirulento, con una mayor incidencia relativa de listeriosis grave aun en pacientes sin comorbilidades conocidas o demostradas16,19. Los CC3 y CC7, en cambio, son considerados clones intermedios entre los mayormente asociados a casos clínicos y los encontrados principalmente en alimentos. Aunque la prevalencia de estos complejos es baja, han sido implicados en brotes en distintas regiones geográficas15,28.
Las muestras de alimentos se correspondieron mayormente con el CC1 (serogrupo IVb) y el CC9 (serogrupo IIc), seguido por el CC3 (serogrupo IIb). El CC9 es un clon hipovirulento con una baja frecuencia clínica y a menudo encontrado en muestras de distintas matrices alimentarias debido a su alta resistencia a distintas condiciones de estrés16. Como una infección surge de la interacción entre el huésped y los factores bacterianos, la ausencia de factores de virulencia específicos de los clones hipovirulentos es compensada por la presencia de comorbilidades significativas o inmunosupresión grave en el huésped, que incrementan la posibilidad de desarrollar la enfermedad. Esto destaca la relación entre la virulencia del microorganismo y los factores predisponentes del huésped en la generación de la invasión y la presentación de formas graves de la infección16,27.
Una limitación de este estudio fue la ausencia de información sobre los antecedentes clínicos y la evolución de los pacientes, lo cual hubiera permitido identificar poblaciones claves o factores de riesgo. En Argentina, la listeriosis ha sido recientemente incorporada al Sistema Nacional de Vigilancia de la Salud (SNVS) como evento de notificación obligatoria18. La obtención de información epidemiológica de forma sistemática y homogénea permitirá un mejor conocimiento de la carga de esta enfermedad en distintas poblaciones de pacientes, así como una estimación de su prevalencia e incidencia y el impacto real en la salud pública.
El análisis filogenómico evidenció la presencia en Argentina de tres clados mayoritarios. El clado A representó el 30,1% de los aislamientos y estuvo representado por el clon hipervirulento CC1 (serogrupo IVb). La mayoría de los aislamientos dentro de este clúster mostraron una posible u ocasional relación epidemiológica entre sí (˃10 SNP)10. Sin embargo, cuatro aislamientos de humanos mostraron una alta relación epidemiológica (1-9 SNP)10, lo que sugiere la presencia de un brote desde el año 2020 y que abarcó las jurisdicciones de Entre Ríos, Buenos Aires y Río Negro. Dado que estos hallazgos son dirigidos por los datos moleculares, destacamos la necesidad de considerar evidencia adicional, como datos epidemiológicos, hábitos alimenticios (mediante encuesta alimentaria) y evidencia de rastreo como parte de la vigilancia de la listeriosis y las investigaciones de brotes. Además, la evaluación de posibles vínculos epidemiológicos debería considerar las distintas etapas de la cadena alimentaria, incluidas las fuentes de contaminación externa, como entornos urbanos y naturales11. Los clados B y C se correspondieron con el CC3 (serogrupo IIb) y el CC9 (serogrupo IIc), respectivamente. En ninguno de estos clados se observó una alta relación epidemiológica entre aislamientos de humanos y alimentos (˂10 SNP)10. Sin embargo, dos aislamientos de humanos del clado B y uno del clado C presentaron una posible relación epidemiológica con aislamientos de alimentos (11-14 SNP en ambos casos)10. El estudio de un mayor número de aislamientos de matrices alimentarias y de las distintas etapas del proceso permitiría trazar una dinámica más clara de la transmisión de L. monocytogenes y, posiblemente, evidenciar aspectos desconocidos de la epidemiología de la listeriosis. Además, la actual falta de datos genómicos de L. monocytogenes de diversas fuentes y regiones geográficas es una barrera para el manejo eficaz de este patógeno, tanto para las autoridades de salud pública como para los actores de la industria alimenticia.
En conclusión, el estudio genómico de la dinámica poblacional de aislamientos de L. monocytogenes en Argentina mostró la presencia de clones circulantes con distintos grados de virulencia. En este conjunto de aislamientos predominó el clon hipervirulento CC1 y serogrupo IVb en muestras de humanos y alimentos. Asimismo, el análisis de SNP evidenció una relación epidemiológica elevada o posible entre aislamientos de humanos y/o de alimentos. Estos hallazgos marcan la necesidad de un fortalecimiento del sistema sanitario en cuanto a la identificación de casos índices, la investigación epidemiológica, los controles bromatológicos y el análisis genómico a fin de elaborar medidas preventivas de promoción y protección mejor dirigidas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Este estudio fue realizado con los fondos de la Administración Nacional de Laboratorios e Institutos de Salud (ANLIS) «Dr. Carlos G. Malbrán» – Ministerio de Salud, Argentina.
