


El síndrome del cráneo en trébol, o síndrome de Kleeblattschädel, es una malformación rara en la cual el cráneo presenta un aspecto de trébol. Es causado por el cierre prematuro de varias suturas, evidenciándose desde antes del nacimiento.
Objetivopresentar nuestra experiencia en un caso de síndrome del cráneo en trébol, y actualizar la información de la literatura.
Caso clínicolactante de sexo femenino, 5 meses de edad, diagnósticos al nacimiento de Fisura labio-Palatina e Hidrocefalia. A los 30 dias de vida se instaló valvula ventrículo peritoneal, y se procedió a enucleación ocular bilateral por proceso infeccioso. Se controla en Genética donde se confirma macrocefalia y craneosinostosis tipo cráneo en trébol. El estudio citogenético 46XX, Ecocardiografía normal, TAC de cerebro mostró anomalias múltiples asociadas a hidrocefalia y malformaciones inespecíficas.
ConclusionEl cráneo en trébol puede presentarse aisladamente o asociado a otras anomalías congénitas, conformando varios síndromes de craneosinostosis, como Crouzon, Pfeiffer o Carpenter. También puede ser componente de la secuencia de rotura amniótica o de diversas displasias, como la campomélica, tanatofórica tipo ii, o la distrofia torácica asfixiante de Jeune. El caso descrito no cumple con todas las características necesarias para incluirlo dentro de un síndrome específico, y no habiendo antecedentes familiares que sugieran patrón de herencia ni anomalías cromosómicas se concluye que se trata de un caso de anomalías congénitas de presentación esporádica.
Cloverleaf skull syndrome, or Kleeblattschädel syndrome, is a rare malformation in which the skull has a cloverleaf appearance. It is caused by the premature closure of several sutures, being evident before birth.
ObjectiveTo present our experience in a case of cloverleaf skull syndrome, and update the information from the literature.
Clinical caseA female infant of 5 months of age, diagnosed at birth with cleft lip and palate and hydrocephaly. A peritoneal ventricle valve was implanted at 30 days of life, and an ocular enucleation was performed due to an infectious process. The patient was followed-up in Genetics, where it confirmed a macrocephaly and craniosynostosis type cloverleaf skull. The 46XX cytogenetic study and echocardiography were normal. The brain CT scan showed multiple anomalies associated with hydrocephaly and non-specific malformations.
ConclusionCloverleaf skull may be present in isolated form or associated with other congenital abnormalities, leading to various craniosynostosis syndromes, such as Crouzon, Pfeiffer or Carpenter. It may also be a component of the amniotic rupture sequence or to different dysplasias, such as campomelic dysplasia, thanatophoric dysplasia type 2, or the asphyxiating thoracic dystrophy of Jeune. The case presented does not fulfil all the characteristics needed to be included within a specific syndrome, and on not having a family history that suggests a hereditary pattern or chromosome abnormalities, it is concluded that it is a case of a congenital anomaly of sporadic presentation.
Lactante menor, de 5 meses de edad y sexo femenino, producto de un tercer embarazo, con parto por cesárea en el Hospital Regional de Lambayeque (Lambayeque, Perú) y peso al nacimiento de 4,010kg. Los diagnósticos al nacimiento fueron fisura labio-palatina (FLP) bilateral e hidrocefalia. A los 30 días de vida se le colocó una válvula de derivación ventrículo peritoneal (DVP) y fue sometida a enucleación ocular bilateral debido a un proceso infeccioso. Al mes y 20 días la paciente es referida al Instituto Nacional de Salud del Niño San Borja (Lima, Perú) para tratamiento quirúrgico de la FLP.
A la edad de 5 meses en la evaluación clínica en el servicio de genética se describe un peso de 8,9kg (Z score: 4,31), talla 68cm (Z score: 3,32), y perímetro cefálico: 47cm (Z score: 4,16). Frecuencia cardiaca 128 latidos/minuto, frecuencia respiratoria 32/min. Al examen físico se evidenció macrocefalia con craneosinostosis tipo cráneo en trébol, además de facies dismórfica con hipotelorismo y fisura facial bilateral que abarcaba desde los cantos internos hasta el labio superior. Ausencia de globos oculares. Presentaba además fisura labio palatina bilateral, fontanela anterior de 1,2cm de diámetro y ausencia de globos oculares y párpados. Tórax y pulmones: murmullo vesicular audible en ambos hemitórax. Cardiovascular: ruidos cardiacos rítmicos, no soplos audibles. Abdomen: blando, depresible, sin visceromegalia ni hernia umbilical palpable, cicatriz operatoria en el flanco derecho. Extremidades: destacaba hipotonía global marcada. Pie derecho con braquidactilia y constricción por brida amniótica en el tercio inferior de la pierna izquierda. Genitales: femeninos normales. Con respecto al desarrollo psicomotor balbuceaba y no presentaba control cefálico.
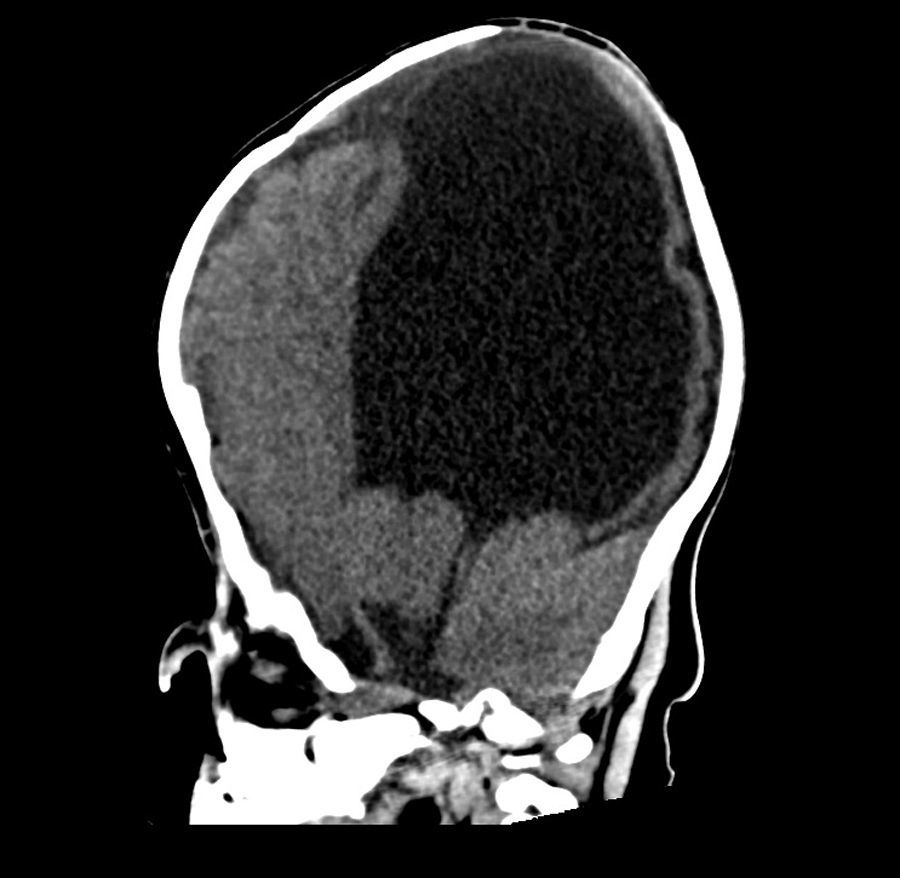
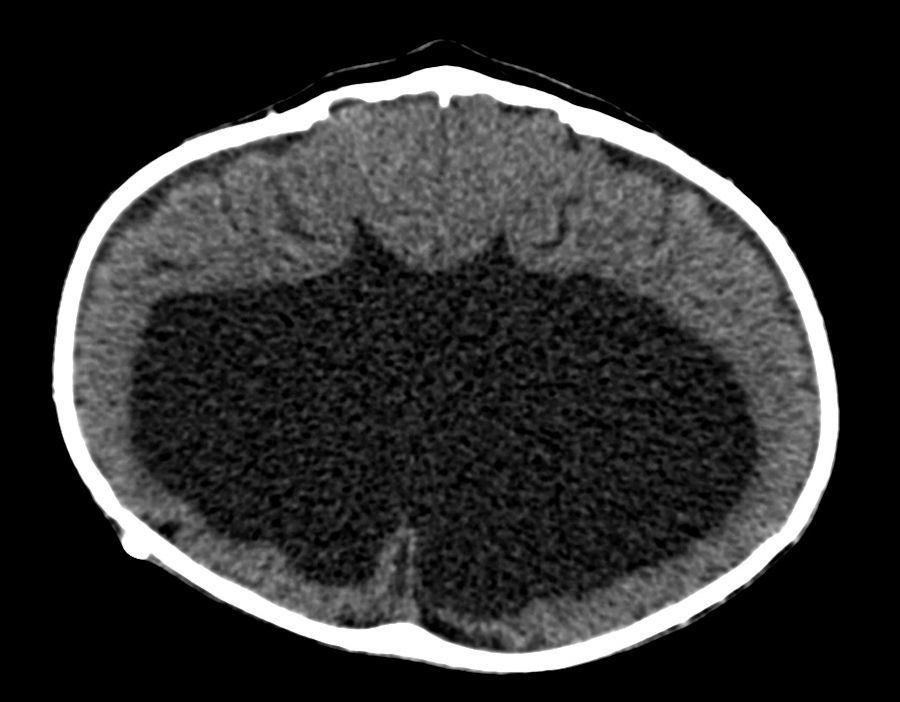
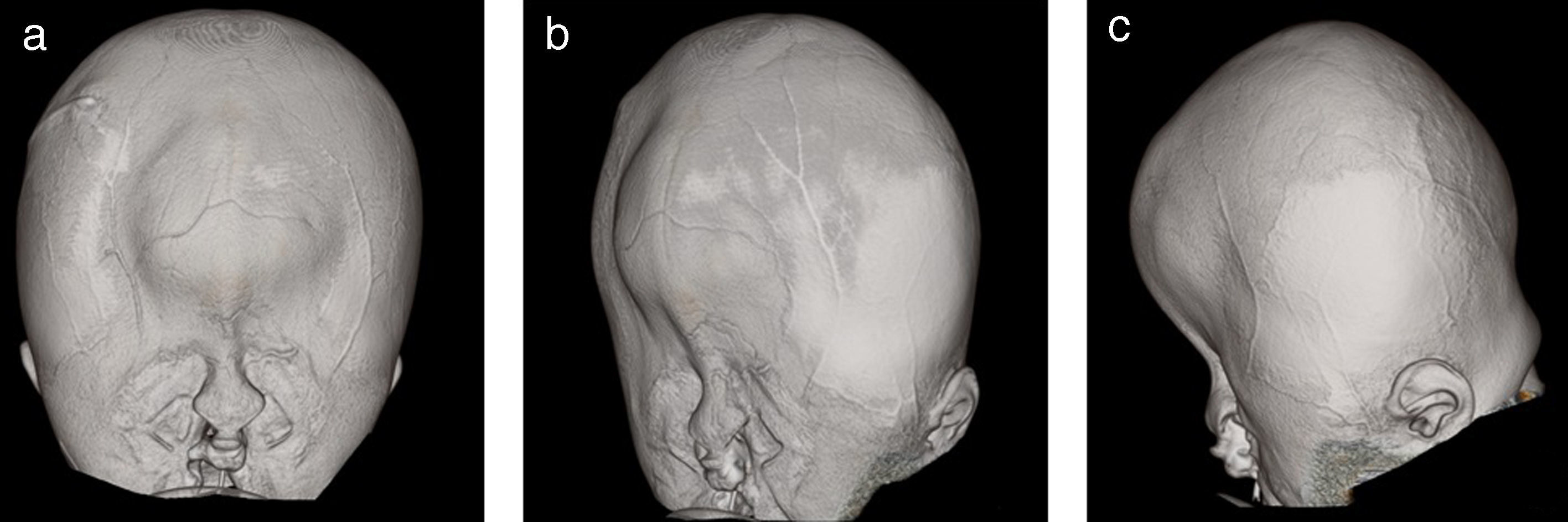
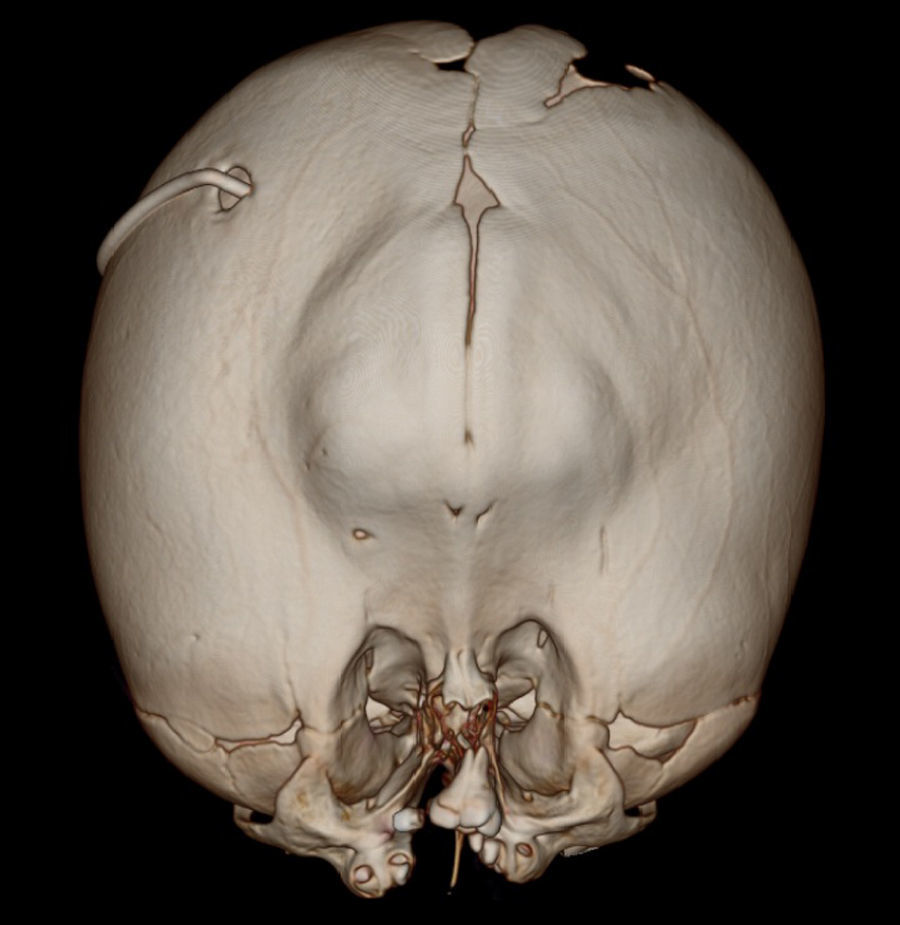
Se le realizaron estudios para descartar otras malformaciones; la tomografía cerebral sin contraste mostró pérdida de volumen del parénquima cerebral, predominio de sustancia blanca de los lóbulos parietales, temporales y occipitales, con atenuación de la diferenciación entre sustancia gris y blanca. Marcada dilatación de los ventrículos laterales, predominio de astas posteriores e inferiores, adelgazamiento marcado del cuerpo calloso, ausencia de septum pellucidum, fusión de ambos tálamos con ausencia de tercer ventrículo. No se definía el acueducto de Silvio y los ganglios basales solo estaban definidos parcialmente. No se evidenció tumoración intraventricular, signos de sangrado ni calcificaciones (figs. 1 y 2). Se observaba catéter de derivación con ingreso frontal derecho y extremo al nivel del cuerpo del ventrículo lateral derecho. En la reconstrucción tridimensional al nivel de la calota craneana se observó fusión de suturas coronales, con incremento del diámetro transversal y disminución del diámetro anteroposterior. Asimismo, al nivel de las órbitas se observó desplazamiento medial en relación con hipotelorismo, con ausencia de globos oculares por antecedente quirúrgico. En las partes blandas se observó solución de continuidad a nivel del labio superior que se extiende bilateralmente a través de apófisis alveolar, entre los dientes incisivos centrales, comprometiendo la apófisis palatina, la lámina horizontal del hueso palatino y el paladar blando hasta su extremo posterior (figs. 3 y 4).


. A. Desplazamiento medial orbitario en relación con hipotelorismo. B. Abombamiento bitemporal asimétrico. C. Herniación frontal.")

Se realizó el estudio citogenético cuyo resultado fue normal, 46,XX. Se realizó una evaluación cardiológica con ecocardiograma, que descartó anomalías. Los potenciales evocados auditivos fueron normales. Actualmente la paciente se alimenta por sonda nasogástrica y se encuentra a la espera del manejo quirúrgico reconstructivo.
DiscusiónLa malformación de cráneo en trébol es una rara1,2 y heterogénea deformidad con diferentes etiologías y grados de severidad3–5. Diversos reportes han sido publicados desde su descripción hace 54 años6, siendo identificada con mayor frecuencia en el género femenino, como en el caso presentado. El diagnóstico puede ser prenatal, mediante estudios de imágenes, en los que se evidencia cráneo trilobular o en trébol con abombamiento bitemporal que puede ser asimétrico, asociado a herniación frontal por un anillo constrictor2. En la etapa posnatal el diagnóstico es clínico, basado en el fenotipo. El síndrome de Kleeblattschädel es considerado clásicamente resultado de la sinostosis intrauterina de la sutura coronal y lambdoidea asociada con hidrocefalia3. El presente caso, como en otros reportados, presenta un patrón inespecífico de sinostosis de las suturas7–9.
En las formas más graves el abombamiento bilateral del cráneo en las regiones temporales y frontoparietales induce un desplazamiento hacia abajo de los pabellones auriculares2,10. Puede haber exoftalmos asociado a hipoplasia orbitaria con imposibilidad de cerrar los párpados, provocando ulceración corneal11 y riesgo infeccioso. El estudio de imágenes del caso evidencia desplazamiento medial orbitario con hipotelorismo (fig. 3). Además, en esta condición se han descrito otros signos dismórficos como aplanamiento de la raíz nasal e hipoplasia mediofacial. También puede asociarse encefalocele, onfalocele y cardiopatías tales como: comunicación interauricular, válvula aórtica bicúspide, persistencia del conducto arterioso, entre otras5,6. Por ello la importancia de la evaluación ecocardiográfica. En el síndrome de Kleeblattschädel se asocia a hidrocefalia, por obstrucción acueductal2; en el caso presentado, ello condicionó la DVP (figs. 1 y 4).
El cráneo en trébol puede presentarse aisladamente o asociado a otras anomalías congénitas, conformando varios síndromes de craneosinostosis, como: Crouzon, Pfeiffer o Carpenter. También puede ser componente de la secuencia de rotura amniótica o de diversas displasias, como la campomélica, tanatofórica tipo ii, o la distrofia torácica asfixiante de Jeune3,12.
El caso descrito no cumple con todas las características necesarias para incluirlo dentro de un síndrome específico, y no habiendo antecedentes familiares que sugieran patrón de herencia ni anomalías cromosómicas se concluye que se trata de un caso de anomalías congénitas de presentación esporádica.
En la mayoría de casos en el síndrome de Kleeblattschädel se produce una muerte temprana4,12. En casos excepcionales de supervivencia hay retraso psicomotor asociado, como en el caso de la paciente. El aumento de presión intracraneal requiere una derivación ventrículo peritoneal2.
ConclusiónEl síndrome de Kleeblattschädel es un tipo de craneosinostosis de etiología genética o ambiental. La asociación de otras anomalías congénitas incide directamente en la evolución y en el pronóstico. El manejo oportuno de la hidrocefalia y de la hipertensión endocraneal subsecuente es determinante, y está estrechamente ligada a la posibilidad de una mayor sobrevida. La corrección quirúrgica agresiva de la craneosinostosis puede mejorar el pronóstico neurológico, aunque en el caso descrito, la asociación con hidrocefalia y fisura facial bilateral condicionan un riesgo elevado de morbimortalidad.
Conflicto de interesesEste trabajo cumple con los requisitos sobre consentimiento/asentimiento informado, comité de ética, financiación, estudios animales y sobre la ausencia de conflicto de intereses según corresponda.
