


La enfermedad granulomatosa crónica (EGC) es una forma infrecuente de inmunodeficiencia primaria que se caracteriza por una sensibilidad anormal a infecciones bacterianas y fúngicas, debida a un déficit en el complejo nicotinamida adenina dinucleótida fosfato oxidasa (NADPH) en los fagocitos.
ObjetivoDescribir tres casos de EGC con énfasis en su forma de presentación y realizar una revisión del tema.
Casos ClínicosSe presentan tres casos clínicos, dos de ellos con relación de parentesco (primos en primer grado). Se llegó a diagnóstico molecular en uno de los casos. Se destacan las manifestaciones clínicas: infecciones recurrentes, abscesos, adenitis y granulomas, y complicaciones, con la finalidad de facilitar la sospecha diagnóstica de EGC, debido a la importancia del diagnóstico temprano y el consejo genético.
ConclusionesLa EGC es un trastorno inmunológico primario congénito infrecuente, con herencia ligada a X en su mayoría, pero también con formas autosómicas recesivas, con una forma de presentación característica y cuyo diagnóstico debe ser oportuno para evitar complicaciones, realizar profilaxis y tratamiento agresivo de las infecciones y consejo genético.
Chronic granulomatous disease (CGD) is a rare form of primary immunodeficiency disease, characterized by an abnormal susceptibility to bacterial and fungal infections, and it is caused by a deficit in the phagocyte nicotinamide adenine dinucleotide phosphate oxidase complex (NADPH), resulting in the inability to generate reactive oxygen species that destroy microorganisms. The diagnosis is based on clinical characteristics and analysis of phagocytes, and later confirmed by molecular studies. Its management should consider antimicrobial prophylaxis, a search for infections and aggressive management of these.
ObjectiveTo describe three cases of CGD emphasizing their forms of presentation and to conduct a review of the condition.
Case reportsThree case reports, two of them first cousins, are presented. Molecular diagnosis was reached in one of the cases. Recurrent infections, abscesses, adenitis, granulomas and complications are identified to facilitate the suspected diagnosis of CGD, bearing in mind the importance of early diagnosis and genetic counseling.
ConclusionsEGC is a rare congenital primary immunodeficiency disorder, mostly with X-linked inheritance, autosomal recessive form, and a specific presentation form. Its diagnosis should be timely to avoid complications. Prophylaxis and aggressive treatment of infections should be performed, as well as genetic counseling.
La enfermedad granulomatosa crónica (EGC) es una inmunodeficiencia infrecuente, con una incidencia de 1 cada 200 000 a 250 000 recién nacidos vivos1. Afecta principalmente a varones, la mayoría de las mutaciones son ligadas a X y las formas autosómicas recesivas ocurren con mayor frecuencia en comunidades con mayor número de matrimonios consanguíneos2.
La EGC se caracteriza por sensibilidad a infecciones recurrentes y severas, bacterianas y fúngicas, con formación de granulomas, debido a la incapacidad de los fagocitos para generar compuestos reactivos de oxígeno, necesarios para la muerte intracelular de microorganismos fagocitados.
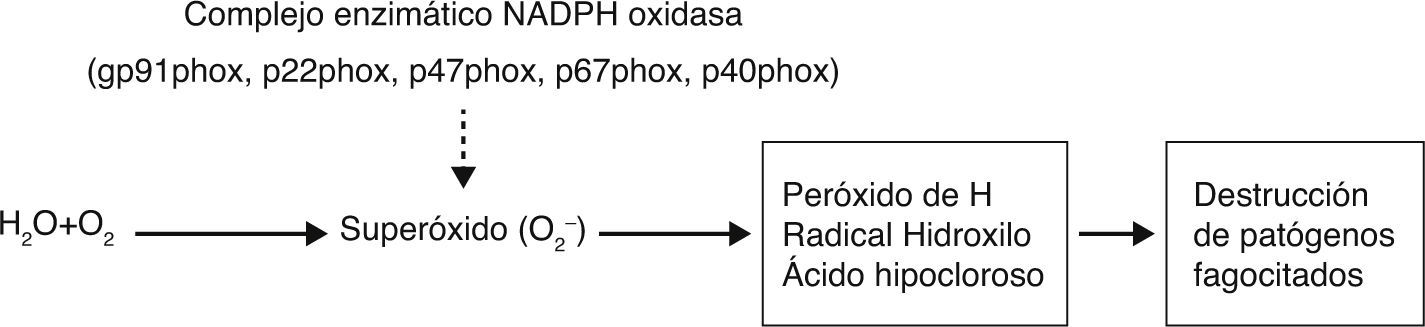
El complejo NADPH oxidasa ensamblado se localiza en la membrana plasmática de los fagocitos. Consta de cinco componentes: dos unidos a la membrana plasmática: proteínas gp91-phox (phagocyte oxidase) y p22-phox que forman el citocromo b558, y tres componentes citosólicos, las proteínas p47-phox, p67-phox y p40-phox3. Este complejo enzimático genera anión súper óxido (O2−), peróxido de hidrógeno (H2O2), ácido hipocloroso (HOCl) y otros productos microbicidas que ejercen efectos tóxicos sobre los microorganismos fagocitados. La estimulación del fagocito, desencadenada por unión de microorganismos opsonizados a receptores en la superficie, conduce al ensamblaje del complejo enzimático activo e inducción del estallido respiratorio (fig. 1)4.
. El O2− se convierte rápidamente en peróxido de hidrógeno, radical hidroxilo y ácido hipocloroso. Estos, junto a los derivados reactivos del nitrógeno y a las enzimas proteolíticas de los gránulos, constituyen el mecanismo fundamental de destrucción de patógenos fagocitados. El defecto de cualquiera de las subunidades del complejo enzimático determina la incapacidad de generar los reactivos de O2.")
Fisiopatología de la EGC por defecto molecular del complejo NADPH.
La NADPH-oxidasa cataliza la transferencia de un electrón desde el NADPH hacia el O2 con la formación de anión superóxido (O2−). El O2− se convierte rápidamente en peróxido de hidrógeno, radical hidroxilo y ácido hipocloroso. Estos, junto a los derivados reactivos del nitrógeno y a las enzimas proteolíticas de los gránulos, constituyen el mecanismo fundamental de destrucción de patógenos fagocitados. El defecto de cualquiera de las subunidades del complejo enzimático determina la incapacidad de generar los reactivos de O2.
La EGC se produce por mutaciones que generan pérdida o inactivación funcional de una de las subunidades del complejo NADPH oxidasa.
Clínicamente, las infecciones y los granulomas son las principales características de EGC5. Los sitios frecuentes de infección son: pulmones, piel, ganglios linfáticos, huesos e hígado. La formación de granulomas es especialmente importante en los tractos gastrointestinal y genitourinario. Las infecciones bacterianas tienden a ser sintomáticas y se asocian con fiebre y leucocitosis; en cambio, las infecciones por hongos se presentan con menos fiebre y menos leucocitosis, lo que dificulta el diagnóstico. Estas a menudo se detectan como hallazgo o en fase avanzada. La respuesta a infecciones virales es normal6. Las localizaciones más frecuentes son: neumonía, abscesos (piel, tejidos, órganos), adenitis supurativa, osteomielitis y bacteriemia/fungemia. Los agentes de la mayoría de las infecciones son Staphylococcus aureus, Burkholderia cepacia, Serratia marcescens, Nocardia y Aspergillus5.
El diagnóstico se realiza por infecciones frecuentes asociadas a granulomas. Frente a la sospecha se deben realizar pruebas de función de neutrófilos. En Chile se encuentra disponible la prueba de nitrobluetetrazolium (NBT), que permite evaluar colorimétricamente la capacidad de los leucocitos de reducir un colorante, y el test de dihidrorodamina (DTH) por citometría de flujo que evalúa la formación de especies reactivas de oxígeno (estallido respiratorio).
El objetivo de esta publicación es describir tres casos de EGC con énfasis en su forma de presentación y realizar una revisión del tema.
Casos ClínicosCaso 1Varón, sin antecedentes mórbidos perinatales. Vacunas al día. Sin antecedentes mórbidos familiares. Presentó infecciones recurrentes: a los 8 meses de edad, neumonía con buena respuesta a amoxicilina, infección del tracto urinario bajo con ecotomografía renal normal; a los 9 meses, adenoflegmón cervical izquierdo drenado quirúrgicamente; a los 18 meses se hospitalizó para estudio de diarrea crónica sin diagnóstico definitivo, que cursó con piodermitis del cuero cabelludo. A los 2 años y 9 meses se hospitalizó por neumonía sin respuesta a tres esquemas antimicrobianos. La tomografía computarizada (TC) de tórax evidenció neumopatía del lóbulo superior derecho y adenopatías hiliares, mediastínicas y retroperitoneales. La fibrobroncoscopia mostró granuloma inflamatorio, se planteó tuberculosis pulmonar como primera opción y se inició tratamiento con isoniazida, pirazinamida, etambutol y cefotaxima, que se suspendió por cultivo de tejido pulmonar positivo para Pseudomonas aeruginosa y Staphylococcus aureus. Aparecieron hepatomegalia y nódulos subcutáneos cuya biopsia evidenció paniculitis flegmonosa. Se inició tratamiento con cloxacilina, amikacina y cotrimoxazol. El estudio inmunológico mostró inmunoglobulinas séricas elevadas, subpoblaciones linfocitarias normales, VIH negativo, PPD 3mm, C3 y C4 normales, test de NBT positivo y test de DTH muy alterado, confirmando diagnóstico de EGC. Se inició profilaxis con trimetoprim/sulfametoxazol e itraconazol. Se realizó estudio molecular que reveló defecto genético homocigoto en gp91-phox, alteración presente también en la madre como defecto parcial. A los 3 y 6 años se hospitalizó por neumonía, sin germen aislado, planteándose infección por Nocardia. A los 7 años presentó osteomielitis de fémur izquierdo con cultivo positivo a Staphylococcus aureus. A los 8 años se hospitalizó por neumonía tratada con voriconazol empíricamente para cubrir Aspergillus fumigatus no confirmado. A los 9 años presentó absceso perianal fistulizado resuelto quirúrgicamente. Mantiene controles ambulatorios sin presencia de infecciones graves, con buen desarrollo pondoestatural. Edad actual, 16 años.
Caso 2Paciente de sexo femenino, sin antecedentes mórbidos perinatales. Antecedentes: en la madre, shock séptico y artritis séptica de cadera por Staphylococcus aureus. Abuela materna con adenoflegmón cervical y celulitis. La tía abuela materna falleció por shock séptico secundario a osteomielitis a los 42 años. Tres hermanos varones sanos. Antecedentes personales de dos episodios de celulitis del cuero cabelludo y dos adenoflegmones cervicales. A los 5 años se hospitalizó por osteomielitis del tobillo derecho y bacteremia por S. aureus que requirió tratamiento quirúrgico y cloxacilina endovenosa, con adecuada respuesta. Exámenes: hemograma con serie roja y serie blanca normales, trombocitosis (476 000 x mm3), IgG e IgM sobre valores normales, IgA normal. Se sospechó EGC confirmada con test DTH con índice de estimulación de 39 alterado (valor normal 138-240). Posteriormente presentó absceso subrotuliano derecho tratado con cloxacilina. Se realizó test de DHR a la madre y la abuela materna, resultando alterado en ambas personas. Mantiene controles en policlínico en profilaxis con cotrimoxazol sin nuevas infecciones. Edad actual, 10 años.
Caso 3Varón de 1 mes y 24 días, sin antecedentes mórbidos perinatales. Primo materno con EGC (caso 1) diagnosticada y confirmada por estudio genético, con madre portadora de mutación de gp91, por lo que se contraindicó vacuna BCG al recién nacido. Se hospitalizó por cuadro febril de 39°C y 24 horas de evolución, con aumento de volumen cervical izquierdo de 1,5cm de diámetro, indurado y móvil, sin signos inflamatorios y sin otros hallazgos relevantes. Exámenes: hematocrito 36%, hemoglobina 11,9 g/dl, leucocitos 33 100 k/μl, 57% segmentados, plaquetas 404 000 K/μl, PCR 25,3 mg/l, sedimento de orina normal, radiografía de tórax normal. Se inició tratamiento con cefotaxima y cloxacilina. La ecotomografía Doppler cervical evidenció en la parte izquierda un conglomerado de adenopatías de aspecto inflamatorio, la mayor de 14 x 7mm. Evolucionó afebril, con descenso de parámetros inflamatorios, hemocultivos y urocultivo negativos, cuantificación de inmunoglobulinas, complemento C3 y C4 normales. Se completó estudio con subpoblaciones linfocitarias normales y test de DTH con índice de estimulación de 2 (rango normal > 50), confirmándose diagnóstico de EGC y se indicó profilaxis con sulfametoxazol-trimetropin. A los 3 meses presentó drenaje espontáneo de la adenitis con regresión progresiva y curación. Último control a los 23 meses con evolución favorable y solo dos episodios de impétigo superficial tratados tópicamente.
DiscusiónSe presentan tres casos de EGC, dos de ellos con relación familiar, con diagnóstico molecular que evidenció mutación de gp91, forma ligada a X2. Según la alteración cromosómica, la EGC puede ser clasificada en ligada a X o autosómica recesiva. De las cinco proteínas que forman el complejo enzimático, la gp91-phox es codificada por el gen CYBB del cromosoma X, su mutación es responsable del 65% de las EGC, y origina la forma ligada a X. Los defectos en los otros componentes, p47, p40 y p67, codificados por genes NCF1, NCF4 y NCF2, respectivamente, producen las formas autosómicas recesivas3,7.
En los tres casos, los sitios de infección más frecuente fueron: tracto respiratorio, en forma de neumonía y granuloma pulmonar; ganglios linfáticos, como adenitis supurativas y alteraciones ganglionares en la TC; y piel, con piodermitis e impétigo. Los casos 1 y 2 presentaron osteomielitis. En el caso 3, la alta sospecha por el antecedente genético, la afectación ganglionar permitió un diagnóstico temprano e iniciar profilaxis antibiótica.
En cuanto a los microorganismos aislados, sólo se logró aislar Staphylococcus aureus en todos los casos y Pseudomonas aeruginosa, y se sospechó neumonía por Aspergillus sin lograr confirmación en el caso 1. En los casos 1 y 2 la osteomielitis por Staphylococcus aureus es concordante con el microorganismo más frecuente. Es importante tener presente como complicación pulmonar por Aspergillus, la “neumonitis de mantillo” (mulch pneumonitis), entidad clínica casi específica en los pacientes con EGC, que se presenta con disnea, hipoxia y fiebre que lleva a insuficiencia respiratoria y muerte después de la inhalación de grandes cargas de esporas e hifas fúngicas, presentes en el heno y el musgo, cuyo rápido reconocimiento es esencial en la supervivencia de los pacientes, pues debe tratarse rápidamente con corticoides y antifúngicos8,9.
Otros organismos aislados con menos frecuencia incluyen especies de Streptococcus, Neisseria meningitidis, Acinetobacter, especies de Candida, Klebsiella pneumoniae, Mycobacterium tuberculosis, micobacterias no tuberculosas, Nocardia, especies de Proteus y especies de Leishmania. No encontramos infecciones por estos microorganismos en estos pacientes, ni tampoco complicaciones por la administración de la vacuna contra la tuberculosis que recibieron los casos clínicos 1 y 2, pero es recomendable no administrar la vacuna como en el caso 3, por los antecedentes familiares10,11.
En el examen físico se puede encontrar hepatomegalia, esplenomegalia o linfadenitis5. Otros trastornos inflamatorios en la EGC se pueden presentar como granulomas, trastornos digestivos, hepáticos, gastrointestinales, genitourinarios, anemia y retardo del crecimiento. Al contrario que en otras inmunodeficiencias, la EGC no se asocia con aumento de neoplasias5. Se pueden presentar, además, cicatrización anormal, diarrea, manifestaciones cutáneas como fotosensibilidad y eczema6. La diabetes, la enfermedad cardiovascular y las nefropatías ocurren con mayor frecuencia en quienes son portadores de la mutación de p4712.
La confirmación molecular se realiza por pruebas de inmunotransferencia, donde se observa ausencia de alguna proteína del complejo NADPH, o por pruebas genéticas para determinar la deleción.
Las piedras angulares del manejo de la EGC son el diagnóstico precoz de las infecciones, el manejo agresivo de las complicaciones infecciosas y la profilaxis antimicrobiana. El uso de cotrimoxazol profiláctico disminuye significativamente la incidencia de complicaciones infecciosas13,14. En los casos 1 y 2, con el inicio de profilaxis, los eventos infecciosos disminuyeron notablemente. Esto es aún más notorio en el caso 3, donde el inicio precoz de profilaxis con cotrimoxazol ha impactado favorablemente la evolución y calidad de vida del paciente. La frecuencia de infecciones bacterianas ha disminuido con el uso de cotrimoxazol como profilaxis. Previamente, la mayoría de las infecciones en pulmón, piel y huesos eran por estafilococos. Actualmente se observan como afectación hepática, ganglionar y de piel, siendo el acné severo facial y la inflamación dolorosa de la mucosa nasal las manifestaciones cutáneas más comunes de infección por Staphylococcus aureus14.
No existen ensayos aleatorios que estudien la profilaxis antibacteriana en pacientes con EGC, pero existen series retrospectivas que sugieren que el cotrimoxazol es eficaz. En caso de alergia a las sulfas, las alternativas son trimetoprim, penicilinas, cefalosporinas o fluoroquinolonas13.
Varias series observacionales y un estudio aleatorizado demostraron que el itraconazol es altamente efectivo como profilaxis antifúngica. En el estudio aleatorio, 39 pacientes fueron asignados a recibir placebo o itraconazol durante un año. Todos los pacientes recibieron profilaxis antibacteriana y la mayoría recibieron profilaxis con interferón gamma (IFN-g). En el grupo placebo se reportaron siete infecciones fúngicas invasivas que necesitaron terapia sistémica, mientras que el grupo que recibió itraconazol reportó solo una infección grave por hongos. La dosis recomendada de itraconazol es de 5 mg/kg de solución oral administrada una vez al día con dosis máxima de 200mg, de por vida15–17.
El tratamiento con IFN-g se ha convertido en parte del manejo en centros de Estados Unidos. Un estudio multicéntrico aleatorizado con IFN-g incluyendo 128 pacientes con EGC con edad media de 15 años, que recibieron IFN-g 50μg/m2 o placebo por vía subcutánea tres veces por semana durante un promedio de 8,9 meses reveló que el 46% de los pacientes del grupo placebo desarrolló al menos una infección grave durante el seguimiento, en comparación con 22% en el grupo de IFN-g. Un año después, el 77% de los pacientes en el grupo de IFN-g aún no había desarrollado una infección grave, mientras que solo el 30% de los pacientes en el grupo placebo estaba libre de infecciones graves. No hubo mejoría en la producción de superóxido por los fagocitos en los pacientes tratados y no se demostró una disminución en las infecciones por Aspergillus18. En nuestro país, su uso está limitado por el costo y se reserva para enfermos con evolución desfavorable a la profilaxis antimicrobiana.
El trasplante de células hematopoyéticas es la cura definitiva para la EGC con mejores resultados en pacientes jóvenes y con menos secuelas, pero también es eficaz en pacientes con infecciones graves recurrentes a pesar de la profilaxis, infecciones difíciles de tratar o enfermedades inflamatorias19–21.
La terapia génica con vectores retrovirales y lentiviral puede reconstituir la actividad de la NADPH oxidasa en las células deficientes, pero no se ha masificado.
El pronóstico ha mejorado gracias al uso de profilaxis con promedio de supervivencia de 40 años. En general, las tasas de supervivencia son mejores en las mujeres que en los hombres, lo cual refleja la mayor gravedad de la EGC ligada a X2–5.
En conclusión, la EGC es una inmunodeficiencia primaria con afectación pulmonar, ganglionar, ósea y de piel, y formación de granulomas. La alta sospecha diagnóstica permite evaluar la función de los fagocitos con test de DHR, e iniciar prontamente terapia antimicrobiana profiláctica con cotrimoxazol e itraconazol, lo que define el pronóstico y la calidad de vida del paciente. El estudio molecular permite, además, identificar el tipo de herencia que presentará la enfermedad, lo que es útil en el consejo genético y en medidas precautorias como suspender la administración de la vacuna contra la tuberculosis, inmunización obligatoria en muchos países.
Conflicto de interésEste trabajo cumple con los requisitos sobre consentimiento/asentimiento informado, comité de ética, financiamiento, estudios animales y sobre la ausencia de conflictos de intereses según corresponda.
