



Las hiperfenilalaninemias se definen por un nivel sanguíneo de fenilalanina sobre 2mg/dl. La principal causa es una mutación en el gen que codifica la fenilalanina hidroxilasa que cataliza la reacción que transforma la fenilalanina en tirosina. Las hiperfenilalaninemias se clasifican en benignas o leves, y las fenilcetonurias en leves, moderadas y clásicas. Debido a que su detección más allá del periodo neonatal causa retardo mental severo, desde 1992 en Chile su detección, junto con la del hipotirodismo congénito, es parte del Programa Nacional de Pesquisa Neonatal. Este artículo pretende responder las preguntas más comunes que se puede hacer el pediatra cuando enfrenta a un paciente con hiperfenilalaninemias.
Hyperphenylalaninaemias are defined by a blood phenylalanine over 2mg/dl. The main cause is due to a mutation in the gene that codes the phenylalanine hydroxylase that catalyses the reaction that converts phenylalanine into tyrosine. The hyperphenylalaninaemias are classified into benign or mild hyperphenylalaninaemias, or mild, moderate or classic phenylketonurias. Due to its delayed detection outside the neonatal period it causes severe mental retardation. Its detection along with congenital hypothyroidism has been part of the National Neonatal Screening Program since 1992 in Chile. This article aims to answer the most common questions asked by the paediatrician when faced with a patient with hyperphenylalaninaemias.
Las hiperfenilalaninemias (HFA) se definen por los niveles sanguíneos del aminoácido esencial fenilalanina (FA) superiores a 2mg/dl.
En 1984 se inició en Chile un plan piloto de pesquisa neonatal de HFA demostrando que era factible implementarlo. En 1992, el Ministerio de Salud comenzó un Programa Nacional de Pesquisa de HFA e hipotiroidismo congénito, extendiéndose a todo el país en 1996. Actualmente cubre el 98,7% de todos los recién nacidos (RN), estableciendo una incidencia de 1:18,916 RN para fenilcetonuria clásica, siendo diagnosticados a los 18,6±9 días1. Para HFA la incidencia es de 1:10,198 RN. Desde 2005 este examen es obligatorio (Ley 19.966 sobre garantías explicitas en salud)1.
El tratamiento nutricional debe iniciarse oportunamente antes de los 30 días de vida para prevenir el retardo mental, y debe mantenerse a lo largo de toda la vida, consistiendo en la restricción del aminoácido FA de la dieta y el uso de una fórmula especial, subvencionada por el Ministerio de Salud hasta los 18 años.
El objetivo de este artículo es responder las preguntas que podría hacerse un pediatra cuando se enfrenta a un niño que ha sido diagnosticado con una HFA en su práctica diaria.
DefiniciónLas HFA corresponden a un conjunto de condiciones caracterizadas por una elevación del aminoácido FA en la sangre definida por un valor>2mg/dl y se definen por una relación entre los aminoácidos FA y tirosina (TIR) persistentemente mayor a 3, utilizándose este valor en la confirmación diagnóstica2.
EtiologíaLas HFA son condiciones genéticas de herencia autosómica recesiva, donde se asume que ambos padres son portadores. El riesgo de recurrencia de la enfermedad es de un 25% en futuros embarazos. Aproximadamente en un 98% de los casos, las HFA se producen por el déficit o ausencia de la enzima FA hidroxilasa codificada en el gen 12q22-q24.1, que cataliza la reacción de FA a TIR. En el 1-2% de los casos la causa es un defecto en el sistema cofactor de esta enzima, tetrahidrobiopterina3,4.
Clasificación¿Hay diferentes tipos de hiperfenilalaninemias? ¿Son todas igual en severidad?Dentro de las HFA existen diferentes grados de severidad, fenómeno debido a la gran cantidad de mutaciones que afectan en diferente medida la actividad de la enzima FA hidroxilasa. Es así como las HFA se pueden clasificar, de acuerdo con el nivel de FA plasmática, la tolerancia a la ingesta de este aminoácido, la actividad enzimática residual y las mutaciones que la originan, en5:
- •
Fenilcetonuria (PKU) clásica (OMIM 261600)
Es la más severa, definiéndose por niveles de FA plasmática sobre 20mg/dl, con TIR menor de 2mg/dl, fenilcetonas en orina, actividad de FA hidroxilasa inferior a un 1% y tolerancia a ingestas de FA menor a 20mg/kg/día (entre 250 y 350mg/día).
- •
PKU moderada
Presentan FA plasmática entre 6 y 19mg/dl, TIR normal, actividad de FA hidroxilasa entre 3 y 50% y tolerancia a ingestas de FA entre 20-25mg/kg/día (350-400mg/día).
- •
PKU leve
Con niveles entre 4 y 6mg/dl, tolerancia a ingestas de FA mayores, entre 25 y 50mg/kg/día (400-600mg/día).
- •
HFA leve
Presentan niveles de FA plasmática entre 2 y 4mg/dl y nivel de TIR normal, actividad de FA hidroxilasa del 50%. Puede normalizar niveles a los 6 meses, recomendándose una dieta normal con supervisión regular de los niveles.
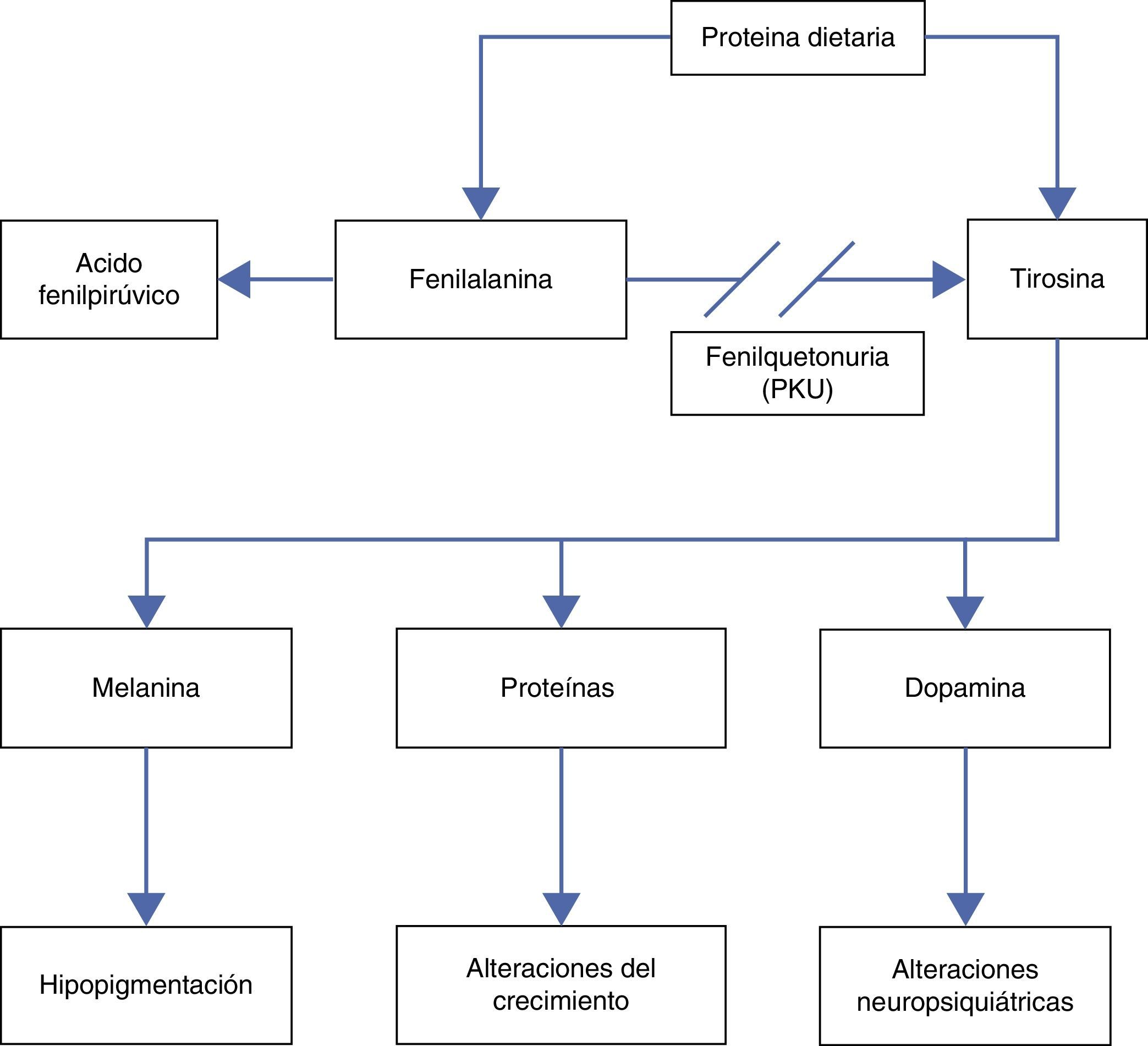
Debido a la deficiente actividad de la enzima FA hidroxilasa, se produce acumulación de FA y, por consiguiente, disminución de la producción del aminoácido TIR (fig. 1).

El aumento de FA en la sangre produce una inhibición competitiva del transporte de otros aminoácidos a través de las membranas celulares, incluyendo el paso por la barrera hematoencefálica, aumentando más la concentración de FA en el cerebro. Lo anterior reduce las concentraciones de aminoácidos intraneuronales e inhibe también competitivamente la hidroxilación de TIR y triptófano, causando una disminución de la síntesis proteica con afección de la proliferación dendrítica, la mielinización e inhibiendo la síntesis de serotonina, dopamina y norepinefrina5. Por otro lado, la deficiencia de TIR explica la afección del crecimiento pondoestatural y los signos de hipopigmentación.
Cuadro clínico¿Cuál es el cuadro clínico de las hiperfenilalaninemias?Afortunadamente, y gracias al programa de pesquisa neonatal, es cada vez más difícil que nos encontremos con un cuadro completo de PKU clásico tal y como fue descrito por primera vez en 1934 por Asbjörn Folling, en que los niños sin tratamiento iniciaban durante los primeros meses de vida desinterés por el medio, convulsiones (frecuentemente espasmos masivos), eczema rebelde al tratamiento y olor a humedad. En casos no tratados oportunamente el retardo del desarrollo se hace evidente en el segundo trimestre de la vida, seguido en la etapa preescolar y escolar por graves trastornos de la conducta, agresividad, hiperactividad, comportamientos autoagresivos y rasgos autistas. En adultos se observaba retardo mental profundo y convulsiones de difícil tratamiento5.
Hoy en día, luego de instaurado el programa de pesquisa, los niños PKU con buen control metabólico son indistinguibles de aquellos que no presentan la enfermedad tanto a nivel intelectual como antropométrico y psicosocial.
Los pacientes con HFA leve o benigna, clásicamente se describen como asintomáticos independientemente del control, sin embargo, estudios realizados durante las últimas décadas han determinado que los pacientes con HFA y mal control metabólico presentan alteraciones cognitivas en estudios neuropsicológicos6,8.
DiagnósticoEl diagnóstico se puede hacer a través de una gota de sangre tomada en papel filtro, el cual a través de la técnica de fluorometría es capaz de cuantificar los niveles de FA en la sangre, método que se utiliza para realizar la pesquisa neonatal. La espectrometría de masas en tándem (perfil de acilcarnitinas) se utiliza para medir la relación FA/TIR y corroborar el diagnóstico si esta es>3mg/dl.
¿Por qué es tan importante el diagnóstico precoz?La PKU clásica conduce a un retardo mental profundo si no es diagnosticada y tratada desde el periodo neonatal. El diagnóstico precoz, antes del mes de vida, y una instauración oportuna del tratamiento permite prevenir el retardo mental y todas las secuelas descritas.
¿Cómo es el procedimiento de pesquisa?A todos los RN, tanto del sistema de salud público como privado chileno, se les extrae una muestra de sangre por punción del talón, la cual se deposita en una tarjeta de papel filtro, siendo analizada mediante la técnica de fluorometría de McCaman y Robins modificada. Si el nivel de FA es mayor a 2mg/dl se informa como positivo y desde el laboratorio del programa (Santiago o Concepción) se solicita inmediatamente la repetición de la muestra, la cual es esta vez analizada por espectrometría de masa en tándem en el Instituto de Nutrición y Tecnología de los Alimentos (INTA) de la Universidad de Chile, examen que es confirmatorio al determinar niveles de FA, TIR y su relación. Con esta confirmación se cita al paciente para una evaluación clínica en el INTA, en donde es ingresado al programa.
¿Qué les ofrece el programa de seguimiento chileno a los PKU?Una vez confirmado el diagnóstico el paciente es ingresado en el Programa de Seguimiento de Enfermedades Metabólicas, el cual se lleva a cabo en el Centro Diagnóstico del INTA. El estado chileno subvenciona exclusivamente la fórmula especial. Los padres deben costear los controles clínicos y los viajes al centro de referencia. Estos pacientes deben acudir regularmente a control con nutricionistas, médico (neurólogo o pediatra) y con psicología, según el calendario preestablecido en el programa.
TratamientoLa base del tratamiento de las HFA consiste en la restricción de FA de la dieta, el uso de una fórmula libre en FA y la suplementación de minerales, ácidos grasos esenciales y TIR en los casos en que se requiera. El seguimiento es clínico (pediátrico, neurológico, psicológico y nutricional) y bioquímico, debiendo durar toda la vida. La dieta excluye todos los alimentos de origen animal y los derivados de los mismos por su alto contenido de FA, y considera los cereales, las frutas y las verduras, cuyo contenido de FA debe ser calculado en forma rigurosa.
Excepcionalmente en casos de HFA leve se podría liberar parcialmente la dieta, pero siempre manteniendo un seguimiento estricto de los niveles plasmáticos de FA. En la tabla 1 se pueden ver los alimentos prohibidos, los controlados y los permitidos.
Alimentos prohibidos, controlados y permitidos en el tratamiento de la fenilcetonuria
| Prohibidos | Controlados | Permitidos libres |
|---|---|---|
| Carnes, pescados, mariscos, huevos, leche y derivados, pan, productos de pastelería, frutos secos, leguminosas, aspartamo y todo alimento que los contenga | Cereales, papas, verduras, frutas, colados infantiles sin alimentos prohibidos | Azúcar, aceite, margarinas, vegetales, jugos de fruta en polvo, maicena, alimentos aproteicos, condimentos |
La fórmula libre de FA cubre los requerimientos de aminoácidos esenciales en estos pacientes, llegando a representar el 80% de la ingesta diaria de proteínas. Esta fórmula es distribuida por el MINSAL a los afectados a través del Programa de Alimentación Nacional Complementaria. Estas fórmulas son de alto costo y actualmente están incluidas en el Programa de Alimentación Nacional Complementaria hasta la edad de 18 años y en mujeres en edad fértil.
En estos pacientes se requiere suplementación de calcio, hierro, cinc y ácidos grasos esenciales para cubrir las deficiencias generadas al excluir de la dieta los alimentos de origen animal. En algunos casos es necesario, además, suplementar con L-TIR.
¿Qué alimentación se debe indicar entre la sospecha y la confirmación diagnóstica?Preferentemente debe mantenerse la lactancia materna. Esto es importante, pues dentro del seguimiento durante el primer año de vida se indica completar el volumen de fórmula especial sin FA con leche materna5.
¿Qué sucede si el paciente o sus padres no siguen las indicaciones dietarias?El objetivo del tratamiento es mantener niveles estables entre 2-6mg/dl durante toda la vida6.
Las consecuencias de una mala adherencia a la dieta van a depender de la edad a la que esta ocurra, la duración del mal control metabólico y la variabilidad de los niveles. Se ha demostrado un menor cociente intelectual con niveles sobre 6mg/dl. El periodo más crítico es obviamente la infancia y la niñez. Sin embargo, se ha demostrado que aun en la adultez, el relajo de la dieta está asociado a una gran variedad de efectos neurocognitivos y psiquiátricos, como déficit en las funciones ejecutivas y síntomas tales como ansiedad, depresión y fobias6–8.
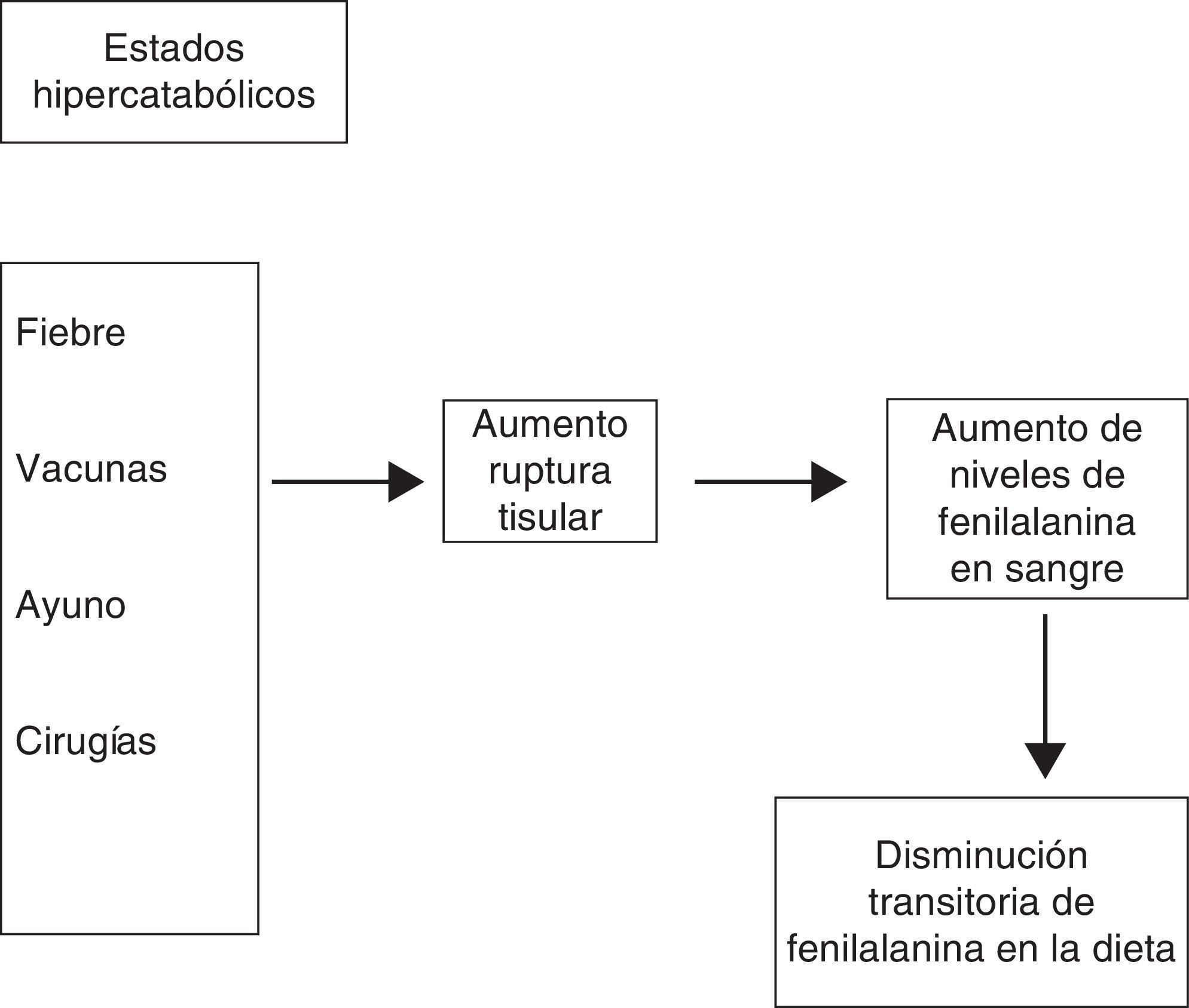
¿Qué hacer si le tocan vacunas?Como todos los niños, los pacientes PKU deben ser vacunados según el Programa Ampliado de Inmunización. Sin embargo, debido a que las inmunizaciones producen catabolismo proteico, debe asegurarse que antes de la vacunación el niño presente niveles plasmáticos de FA menores a 3mg/dl.
¿Qué hacer si el niño PKU se enferma?Cualquier infección constituye un estado hipercatabólico, más aún si genera anorexia o dificultades en la alimentación, elevando, como consecuencia, los niveles de FA. Se debe bajar el aporte de FA de la dieta, para lo cual debe comunicarse con el equipo tratante. Dependiendo del nivel de compromiso que presente el paciente puede requerirse la suspensión total del aporte de FA y mantenerse exclusivamente con fórmula libre de esta, aumentando paralelamente el aporte de calorías para compensar el estado hipercatabólico.
¿El paciente con hiperfenilalaninemia puede recibir cualquier medicamento?En general pueden recibir cualquier medicamento (antipiréticos, antiinflamatorios, antibióticos), evitando jarabes endulzados con aspartamo, los cuales tienen FA.
¿Qué hacer frente a una cirugía?Lo primero que hay que saber es cuántas horas de ayuno requiere la cirugía. Si se puede suspender el ayuno antes de 4-6h, se realimentará con fórmula especial. Si el ayuno es de más de 6h debe mantenerse el suero glucosado al 10%, y si es mayor a 8h se deben agregar lípidos. La realimentación debe ser siempre con fórmula especial (fig. 2).
¿Qué es el síndrome de PKU materno?
Consiste en el fenómeno teratogénico de la FA en el feto de una mujer con HFA. Dado que en la circulación fetal los niveles de FA son unas 1,5 a 1,8 veces más elevados que en la circulación materna, existe un alto riesgo de sufrir malformaciones, como bajo peso al nacer, microcefalia, cardiopatía congénita, retardo mental y retardo del crecimiento intrauterino si es que no existe un buen control metabólico materno. Como el efecto teratogénico más severo suele ocurrir en la primeras semanas de gestación, incluso antes de detectarse un atraso menstrual, es altamente recomendable que todo embarazo en una paciente PKU sea cuidadosamente planificado. La mejor medida de prevención es mantener un control metabólico estricto en toda mujer en edad fértil y trabajar precozmente en educación sexual9.
ConclusiónLas HFA son el error innato del metabolismo más frecuentemente reconocido, con una incidencia de 1/10.000 RN vivos. En Chile tenemos un Programa de Pesquisa Nacional desde el año 1992. Desde entonces su tratamiento y seguimiento a nivel nacional se realiza en el INTA. El pediatra debe conocer esta enfermedad, saber acerca de su programa de pesquisa, familiarizarse con estos pacientes, sobre todo en cuanto a situaciones habituales de la vida de ellos, como periodos de vacunación, infecciones y procedimientos quirúrgicos, así como riesgo de embarazo.
Conflicto de interesesEste trabajo cumple con los requisitos sobre consentimiento/asentimiento informado, comité de ética, financiación, estudios animales y sobre la ausencia de conflicto de intereses según corresponda.
