


El síndrome de regresión caudal es una malformación congénita poco frecuente que abarca un gran espectro de formas de presentación. Caracterizado por el compromiso musculoesquelético caudal, se puede asociar a defectos neurológicos, gastrointestinales, renales y genitourinarios. Aunque su etiología aún no se encuentra aclarada, se ha asociado a la presencia de diabetes materna y a mutaciones en el gen homeobox HBLX9. Su diagnóstico se basa en un buen estudio ecográfico prenatal, un examen físico detallado y estudio imagenológico posnatal con radiografía y/o resonancia magnética. El síndrome de regresión caudal requiere un manejo multidisciplinario en el cual el control metabólico de la diabetes gestacional constituye la mejor medida preventiva disponible en la actualidad.
Se presenta el caso e imágenes de un recién nacido de término de sexo masculino, hijo de madre diabética pregestacional con mal control metabólico y diagnóstico ecográfico prenatal de malformación de columna lumbar, huesos iliacos y extremidades inferiores. Nace en buenas condiciones y se confirma el diagnóstico de síndrome de regresión caudal mediante estudio radiográfico y resonancia magnética compatibles.
Caudal regression syndrome is an uncommon congenital malformation that includes a wide spectrum of clinical presentations. Characterised by caudal musculoskeletal compromise, it can be associated to neurological, gastrointestinal, renal and genitourinary defects. Although the specific aetiology has not been clarified, it has been associated with the presence of maternal diabetes and mutations in homeobox gene HBLX9. Its diagnosis is based on a good prenatal ultrasound detection, detailed physical examination, and post-natal imaging study using radiography and magnetic resonance. Caudal regression syndrome requires multidisciplinary management, and it seems that good metabolic control of gestational diabetes constitutes the best preventive measure available.
We present the clinical case and images of a male term newborn, born to a pregestational diabetic mother with poor metabolic control and a prenatal ultrasound diagnosis of lumbar spine, iliac bones and lower limbs malformation. Born in good conditions, the diagnosis was confirmed using X-rays and magnetic resonance.
El síndrome de regresión caudal (SRC), también conocido como síndrome de agenesia sacra y displasia caudal, corresponde a una malformación congénita severa poco frecuente, cuya extensión clínica va desde agenesia de coxis aislada detectada solo como hallazgo radiológico y sin secuelas, hasta una agenesia sacra o lumbosacra con afectación de los segmentos de médula espinal correspondientes, con malformaciones musculoesqueléticas de la pelvis y extremidades inferiores1. También se han descrito anormalidades neurológicas, ortopédicas, gastrointestinales, genitourinarias y cardiacas2.
Fue Bernard Duhamel3 quién acuñó el término de SRC en el año 1961, para hacer referencia a un espectro de malformaciones que iban desde el ano imperforado hasta la sirenomelia, dos extremos de un síndrome que abarcaba defectos malformativos embrionarios de la región caudal.
La etiología de este síndrome no está esclarecida aún, pero se cree que habría un defecto, antes de la cuarta semana de gestación, en la inducción de los primordios caudales del embrión, y que la diabetes materna jugaría un rol importante en su patogénesis4.
El objetivo de esta comunicación es presentar un caso clínico de un SRC con agenesia lumbrosacra y dismorfia de columna dorsal nacido en la Unidad de Neonatología del Hospital San Borja Arriarán, su diagnóstico presuntivo prenatal y las características radiológicas de este paciente.
Caso clínicoMujer de 44 años, multípara de tres, con antecedentes de diabetes tipo 2 en control y tratamiento desde el año 2011 con glibenclamida. Embarazo actual controlado en alto riesgo obstétrico desde las 12 semanas de gestación, donde inicia desde las 14 semanas tratamiento con metformina e insulina NPH por mal control metabólico y metildopa por síndrome hipertensivo del embarazo.
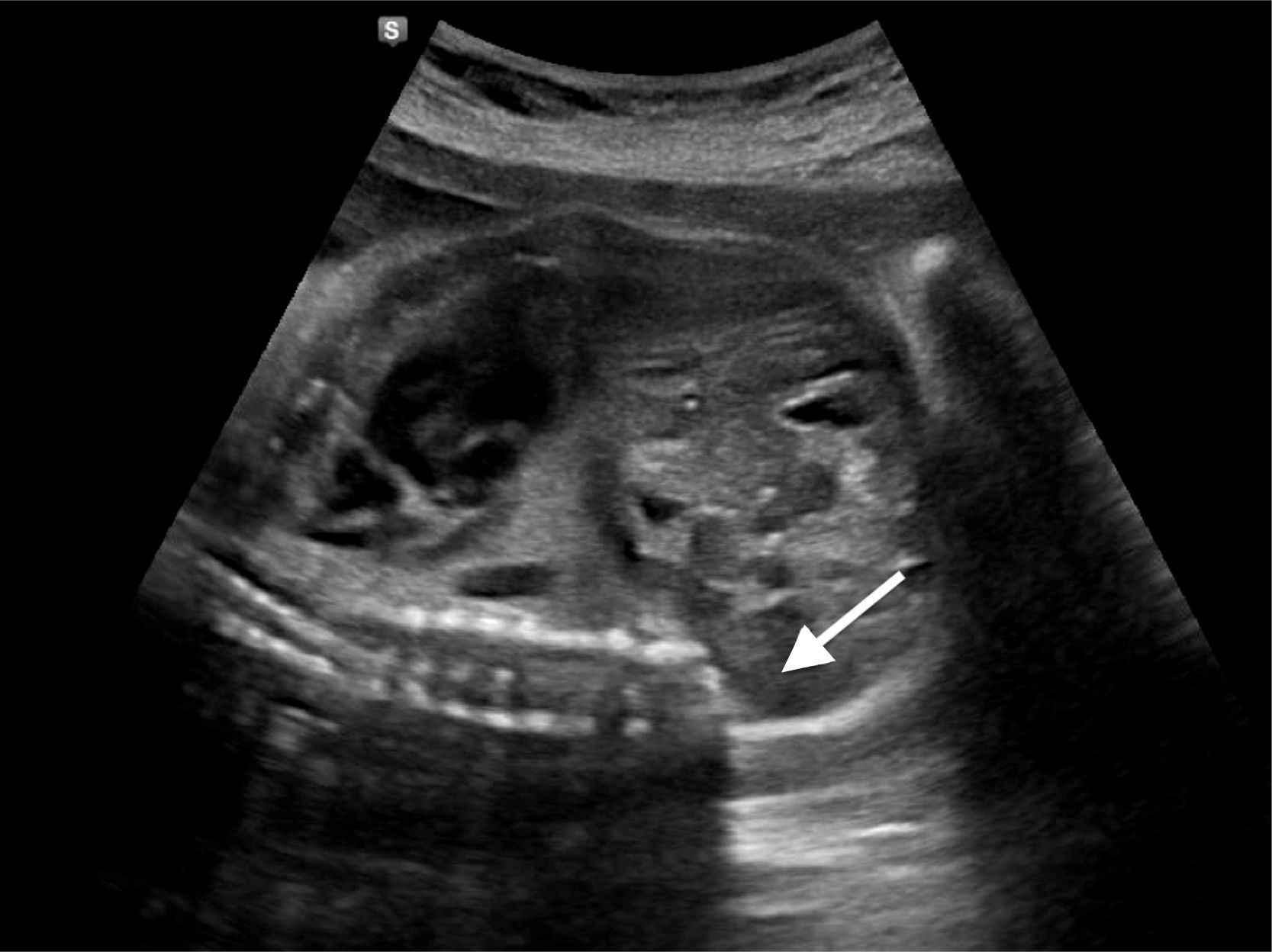
En control ecográfico realizado a las 22 semanas, se detecta malformación a nivel de columna lumbar, ausencia de huesos pélvicos, riñón único y pie equino bilateral; altamente sugerente de síndrome de regresión caudal (fig. 1).
 Agenesia caudal parcial.")
Por rotura prematura ovular de doce horas de evolución, se realiza cesárea de urgencia. Se confirma recién nacido de sexo masculino de 38 semanas de edad gestacional PEG, APGAR 9/9, con un peso de nacimiento de 2.570 g, talla de 45cm y perímetro cefálico 34cm; que evoluciona asintomático en periodo de recién nacido inmediato.
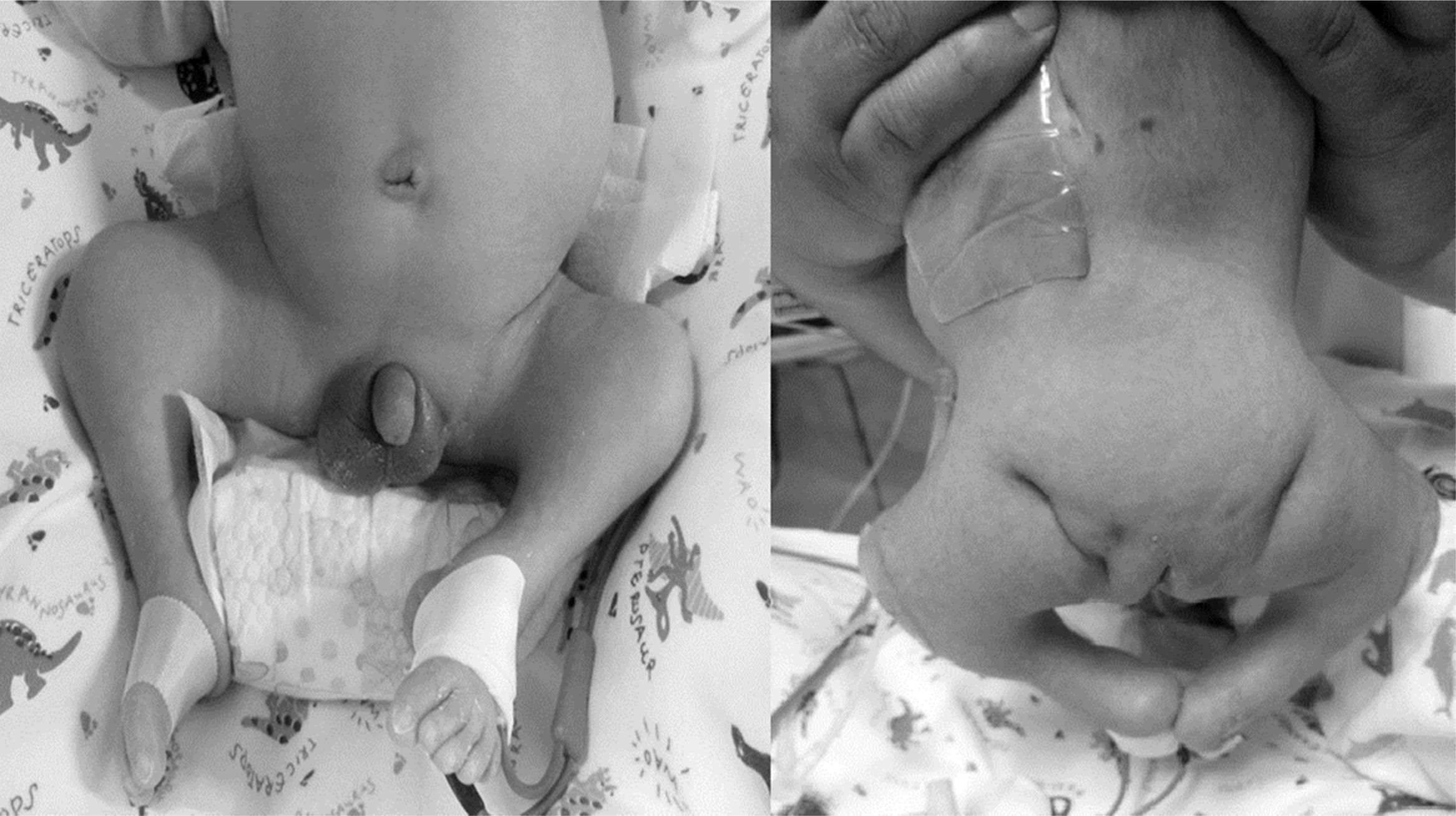
Al examen físico del neonato destaca hipoplasia de hemicuerpo inferior, con extremidades inferiores en flexoabducción, pie equino bilateral y aumento de volumen de partes blandas en fosa iliaca derecha. A la palpación, se detecta ausencia de columna lumbosacra, con genitales masculinos normales (fig. 2). Desde el punto de vista neurológico destaca paraplejia flácida bilateral y ausencia de reflejos osteotendíneos en extremidades inferiores.

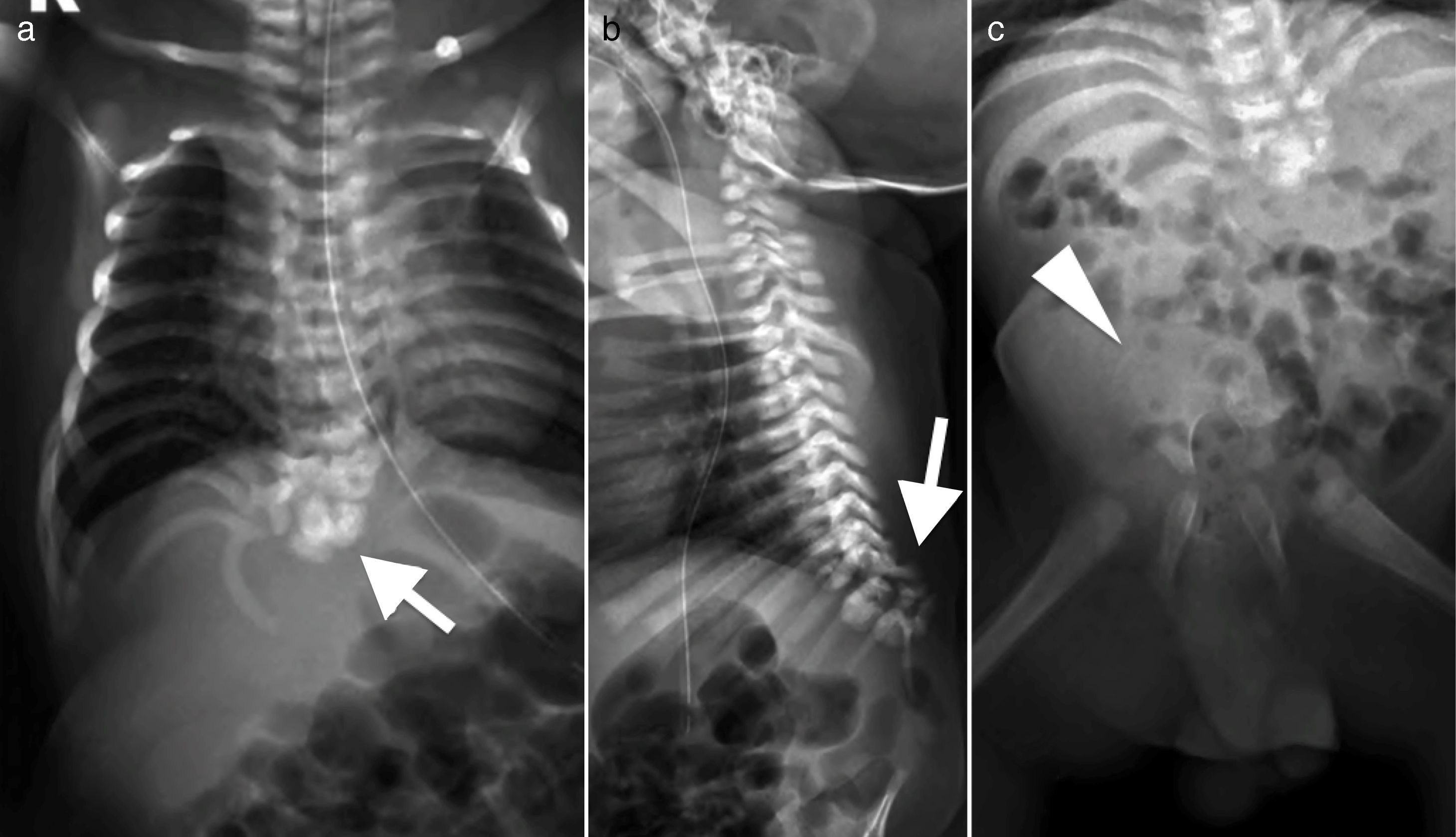
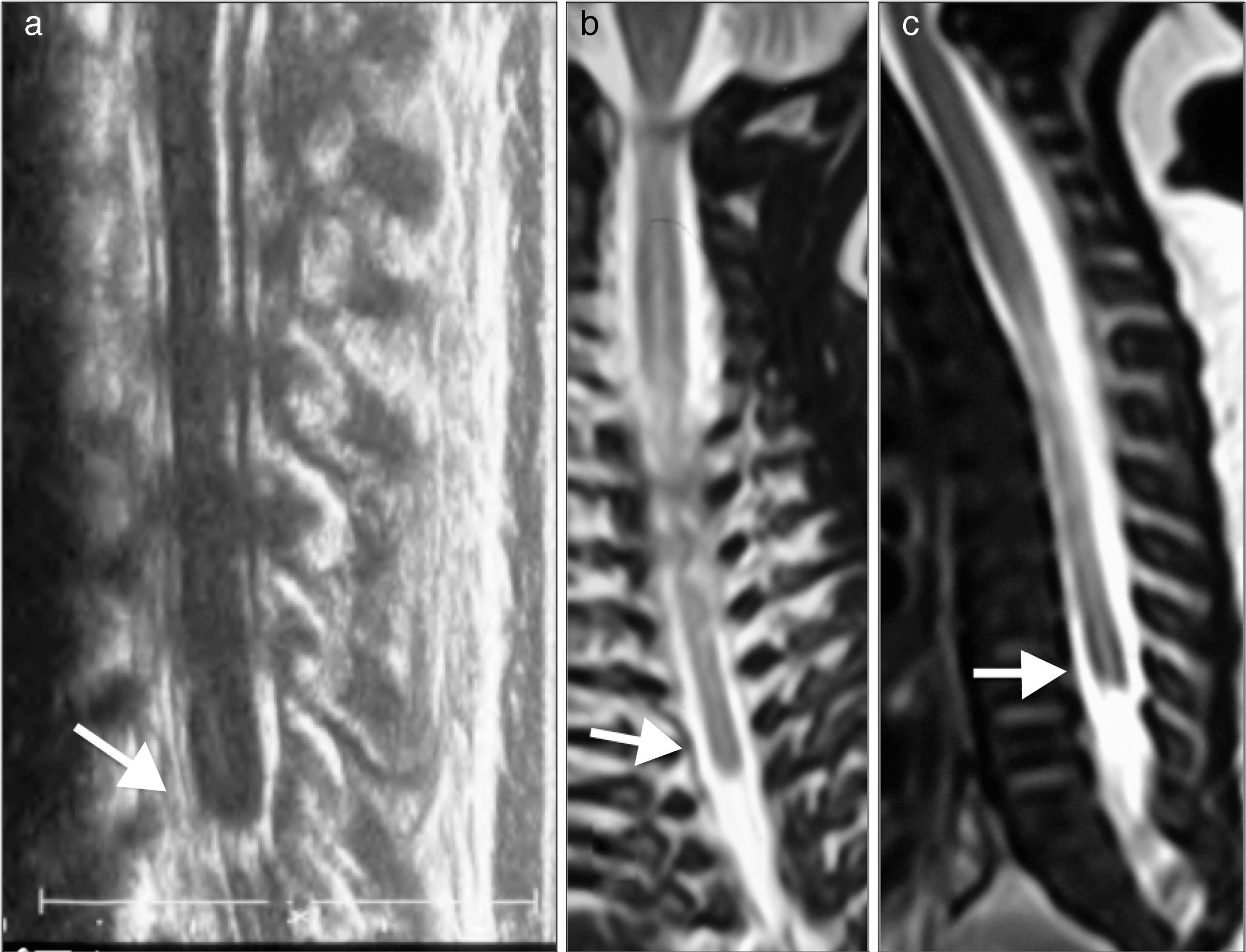
En estudio imagenológico, se realiza radiografía de columna total que confirma interrupción de la columna a nivel dorso distal con ausencia de columna lumbar y sacro, sin continuidad de esta con la pelvis. Pelvis dismórfica con huesos iliacos que se afrontan en línea media en ausencia de sacro (fig. 3). Para complementar estudio, se efectúa resonancia magnética que confirma una malformación espinal compleja, caracterizada por agenesia de sacrocoxis y segmentos lumbares de columna vertebral, dismorfia de cuerpos dorsales bajos (desde D9) con fusión de segmentos distales. La médula espinal presenta su extremo en porción alta (D7) con morfología anormal en forma de «porra» o «nabo». Este aspecto característico del extremo distal de la médula fue también visualizado con ecografía (fig. 4).
 AP. b) Lateral. c) Pelvis. Disgenesia lumbosacra con ausencia total de región lumbar y sacro. Se observa la interrupción de la columna a nivel dorsal bajo (flechas). La pelvis muestra huesos iliacos unidos en línea media sin sacro (cabeza de flecha). En figura c) se hace evidente falta de continuidad y desalineación entre columna y pelvis.")
Radiografía de columna total. a) AP. b) Lateral. c) Pelvis. Disgenesia lumbosacra con ausencia total de región lumbar y sacro. Se observa la interrupción de la columna a nivel dorsal bajo (flechas). La pelvis muestra huesos iliacos unidos en línea media sin sacro (cabeza de flecha). En figura c) se hace evidente falta de continuidad y desalineación entre columna y pelvis.
 Ecografía. b) y c) RM. Médula espinal con extremo distal en situación alta (D7). La morfología del extremo distal medular es anómala y característica de síndrome de regresión caudal, sin el normal ensanchamiento del cono medular, su extremo muestra un término brusco descrito como en cuña, forma de porra o navo (flechas).")
a) Ecografía. b) y c) RM. Médula espinal con extremo distal en situación alta (D7). La morfología del extremo distal medular es anómala y característica de síndrome de regresión caudal, sin el normal ensanchamiento del cono medular, su extremo muestra un término brusco descrito como en cuña, forma de porra o navo (flechas).
El SRC o secuencia de displasia caudal es una malformación poco frecuente, con casos esporádicos cuya frecuencia en nuestro país se desconoce. Según reportes, se observa una incidencia de 0,1-0,25 casos por cada 10.000 embarazos normales, siendo esta mayor en hijos de madres diabéticas en comparación a las no diabéticas, presentándose en uno de cada 350 hijos de mujeres diabéticas, lo que resulta en una incidencia 200 veces mayor en comparación a la población general1,5. El aumento mundial de la DM y embarazo es un reflejo del aumento epidémico de la obesidad6, pudiera ser probable que si hay una relación causal, el SRC se observe con mayor frecuencia en el futuro.
Su etiología no es totalmente conocida en la actualidad, pero se han propuesto un componente genético, la diabetes materna, la hipoperfusión vascular y factores bioquímicos como posibles causas del síndrome. Estudios recientes plantean una contribución, al menos parcial del gen «homeobox» HLXB9, responsable de la morfogénesis y estructura del eje corporal, en la aparición de algunos tipos de agenesia sacra, el cual además se expresa en el páncreas, lo que pudiera vislumbrar la relación entre el SRC y la diabetes7,8. Se le atribuye a la diabetes mellitus la mayor importancia en el desarrollo de la enfermedad. La contribución de la diabetes al desarrollo de malformaciones mayores está bien documentada, y el SRC constituye la anormalidad fetal más característica de la embriopatía diabética9. El mal control metabólico de la glucemia materna al momento de la concepción y durante los primeros meses de la gestación se ha asociado a una mayor incidencia de malformaciones congénitas, teniendo un riesgo 2 a 4 veces mayor10, siendo el desorden del metabolismo de hidratos de carbono crítico durante la diferenciación del mesénquima precartilaginoso, predisponiendo al feto a malformaciones esqueléticas11. La hiperglucemia durante la gestación altera el metabolismo lipídico generando un exceso de moléculas de oxígeno con alta capacidad oxidativa que activa la apoptosis celular, desencadenando las malformaciones fetales12.
Una disminución de mioinositol y de ácido araquidónico también contribuiría a la patogénesis de esta enfermedad debido a que niveles altos de glucemia y glucosa intracelular causarían una disminución en la absorción de mioinositol por parte de los tejidos y del embrión13.
El mecanismo fisiopatológico no ha sido completamente establecido, pero se piensa que habría, antes del día 28 del periodo de gestación, un defecto en la inducción de los componentes caudales del embrión. La alteración ocurriría en la porción medioposterior del mesodermo axial (encargado del desarrollo de cartílago, músculo, esqueleto, dermis, aparato excretor y gónadas) que provocaría ausencia en el desarrollo del brote mesoblástico caudal9. En los avances de la compresión del patrón del mesodermo axial durante las primeras etapas de la gestación se revela que uno o más procesos de migración de la estría primitiva, neurulación primaria o secundaria o la diferenciación están comprometidas. Además, se plantea el tiempo límite de las 4 semanas debido a que hasta esa fecha es cuando la neurulación secundaria completa los segmentos caudales pendientes que no se forman a partir de la neurulación primaria14.
Como existe una relación e interdependencia en el desarrollo de estructuras nerviosas caudales, de la médula, el intestino posterior y los elementos mesonéfricos involucrados en el cierre del tubo neural, resulta en la asociación de anormalidades conjuntas entre el sistema musculoesquelético, neurológico, digestivo, y genitourinario9,15. Así mismo, es infrecuente el compromiso de otras estructuras cuyo desarrollo ocurre distal a los elementos anteriores, como el cerebro, columna vertebral y médula espinal proximal.
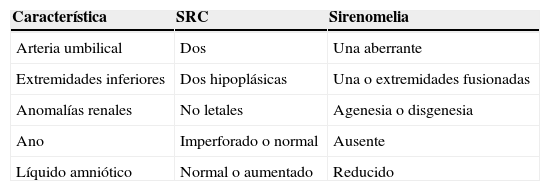
Como se planteó en la introducción, Duhamel definió al SRC como un espectro, en cuyo extremo se encontraría la sirenomelia como máxima expresión de gravedad, pero algunos autores creen que la sirenomelia sería una entidad de patogenia distinta al SRC. A diferencia de este último, la sirenomelia raramente estaría asociada a diabetes materna y se piensa que sería producto de un patrón vascular anormal que condicionaría una isquemia severa del hemicuerpo inferior debido a un vaso aberrante que nace de la arteria vitelina y que se constituye como una sola arteria umbilical16, hallazgo frecuente en estos pacientes, que surge en una porción alta de la aorta abdominal hacia la placenta, desviando el flujo sanguíneo lejos de la región caudal, lo que conduciría a su subdesarrollo15,17. Así fue como Twickler et al., al estudiar 4 casos de sirenomelia y 3 de SRC, describió las pistas que pueden diferenciar a la sirenomelia del SRC: arteria umbilical única aberrante, extremidades inferiores fusionadas, agenesia o disgenesia renal, ano ausente y oligohidroamnios2,18 (tabla 1).
Diferencias entre SRC y sirenomelia
| Característica | SRC | Sirenomelia |
|---|---|---|
| Arteria umbilical | Dos | Una aberrante |
| Extremidades inferiores | Dos hipoplásicas | Una o extremidades fusionadas |
| Anomalías renales | No letales | Agenesia o disgenesia |
| Ano | Imperforado o normal | Ausente |
| Líquido amniótico | Normal o aumentado | Reducido |
Fuente: Zaw et al.15.
El diagnóstico del SRC está basado en los hallazgos ecográficos. Estos son posibles de encontrar ya desde el primer trimestre, por medio de ecografía transvaginal, la que tiene una alta resolución a partir de la semana 17. Un diagnóstico certero es difícil de plantear en el primer trimestre debido a la osificación incompleta del sacro, pero es posible identificar signos ecográficos tempranos como una menor longitud craneocaudal y apariencia anormal del saco vitelino16. Ya en el segundo trimestre, cuando se completa la osificación del sacro, aparecen las anormalidades que varían desde alteraciones del sacro hasta su ausencia y de la columna lumbar, pudiendo asociarse a alteraciones de las extremidades inferiores como deformidad en los ángulos de flexión y en la angulación de los pies19. Un hallazgo típico es encontrar al feto en posición de «Buda» o «rana», cuando mantiene las extremidades inferiores en triple flexión, asociado a otros hallazgos como oligohidroamnios o agenesia renal14. El estudio en el periodo posnatal, tanto para el diagnóstico como para evaluación de la extensión, puede realizarse con ecografía y radiografía de columna total y de extremidades, las cuales evidencian las anormalidades esqueléticas7. El estudio debe completarse con resonancia magnética para evaluar la extensión y compromiso de los tejidos blandos y vísceras20.
Como se expuso anteriormente, el SRC es un espectro clínico, con manifestaciones clínicas que son variables. Ya a la inspección encontramos los primeros estigmas del síndrome, dentro los cuales se puede apreciar una prominencia ósea en espalda correspondiente a la última vértebra, glúteos achatados con aplanamiento del surco interglúteo que acompaña a la agenesia sacra. Las anormalidades ortopédicas son llamativas, dentro de las que se pueden encontrar luxación de caderas y otras alteraciones de la pelvis, deformidades en los pies como pie zambo, contracciones en flexión de rodilla y cadera, defectos en fémur, tibia y fíbula. La escoliosis es la deformidad en columna más frecuente. Dentro del compromiso de otros órganos, las deformidades genitourinarias incluyen malformaciones renales (agenesia y displasia renal, anormalidades ureterales y vesicouretrales) y agenesia y duplicación del conducto de Müller17. En cuanto a los déficits neurológicos, varían desde un mínimo déficit a paresia motora y anestesia sensitiva con atrofia de segmentos inervados por las raíces nerviosas comprometidas a distal de las vértebras alteradas21. A menudo, estas alteraciones se acompañan de vejiga neurogénica. En nuestro caso, muchas de estas alteraciones estaban presentes al examen físico inicial, concordantes con un SRC.
El manejo de estos pacientes es siempre un desafío multidisciplinario, y los fisioterapeutas juegan un rol fundamental. El tratamiento dependerá del grado y localización de la lesión en columna y la existencia de anormalidades asociadas22. Como la enfermedad primaria es intratable en la actualidad, los esfuerzos van dirigidos al tratamiento de soporte. Especial atención requieren las anormalidades ortopédicas y los problemas urológicos, cuyo manejo debe ser abordado precozmente. Estos últimos para evitar las infecciones del tracto urinario a repetición y sus consecuencias como pielonefritis e hidronefrosis, con el consecuente deterioro de la función renal.
En conclusión, el SRC es un raro e infrecuente síndrome malformativo, que se observa predominantemente en hijos de madres diabéticas con mal control metabólico. Por lo mismo, en esta población de riesgo se justificaría un control ecográfico temprano a fin de detectar precozmente la ocurrencia de malformaciones mayores en el feto. El enfoque actual debe ir destinado a la prevención de la diabetes materna, un control adecuado en pacientes que ya sufren la enfermedad y una atención oportuna de la gestación y parto.
Conflicto de interesesEste trabajo cumple con los requisitos sobre consentimiento/asentimiento informado, comité de ética, financiación, estudios animales y sobre la ausencia de conflicto de intereses según corresponda.
