



El síndrome de Sotos (SS) es una enfermedad genética con un patrón de herencia autosómico dominante, causado por haploinsuficiencia del gen NSD1 secundaria a mutaciones puntuales o microdeleciones del locus 5q35 en el que está ubicado el gen. Es un síndrome poco frecuente, presentándose en 7 de cada 100.000 nacimientos. El objetivo de este reporte es presentar el caso de una paciente de 4 años con retardo global del desarrollo, y hallazgos físicos especiales que sugerían un sindrome genético.
Caso clínicoPaciente de 4 años, género femenino, cabello ralo, fascie triangular, fisura palpebral alargada, papadar ojival, mandíbula prominente, escápula alada y clinodactilia del quinto dedo de ambas manos. La prueba molecular de hibridación genómica comparativa por microarreglos, mostró microdeleción de la región 5q35.2 q35.3 de 2.082 MB, que incluye el gen NSD1.
ConclusiónProponemos realizar la prueba de hibridación genómica comparativa en pacientes con retraso global del desarrollo y hallazgos fenotípicos menores.
Sotos Syndrome (SS) is a genetic disease with an autosomal dominant pattern caused by haplo-insufficiency of NSD1 gene secondary to point mutations or microdeletion of the 5q35 locus where the gene is located. It is a rare syndrome, occurring in 7 out of every 100,000 births. The objective of this report is to present the case of a 4 year-old patient with a global developmental delay, as well as specific physical findings suggesting a syndrome of genetic origin.
Clinical caseFemale patient, 4 years of age, thinning hair, triangular facie, long palpebral fissure, arched palate, prominent jaw, winged scapula and clinodactilia of the fifth finger both hands. The molecular test comparative genomic hybridisation test by microarray was subsequently performed, with the result showing 5q35.2 q35.3 region microdeletion of 2,082 MB, including the NSD1 gene.
ConclusionFinally, this article also proposes the performing of comparative genomic hybridisation as the first diagnostic option in cases where clinical findings are suggestive of SS.
El síndrome de Sotos (SS) es una enfermedad genética con un patrón de herencia autosómico dominante, causado en un 90% de los casos por una haploinsuficiencia del gen NSD1 (nuclear proteína SET dominio de unión a receptor-1), con locus 5q35; la prevalencia reportada es 1:15.0001,2. Los pacientes afectados se caracterizan por un crecimiento prenatal y posnatal excesivo, macrocefalia, fascies específicas y retraso en el desarrollo motor, cognitivo y social3. El diagnóstico de SS se confirma con pruebas moleculares que evidencien mutaciones capaces de afectar al funcionamiento del gen NSD1 o deleciones que incluyan la región 5q35.
En pacientes con retardo mental inexplicado y hallazgos fenotípicos inespecíficos, y con el objeto de encontrar la etiología, se ha sugerido realizar hibridación genómica comparativa por microarreglos (HGCm); con esta prueba se examina el genoma del individuo permitiendo detectar alteraciones cromosómicas numéricas y estructurales, pérdida de material genético (como en el SS), o exceso del mismo con un nivel de resolución hasta 1.000 veces mayor que el diagnosticado por cariotipo4.
El objetivo de este reporte es presentar el caso de una paciente de 4 años con retardo global del desarrollo, junto con hallazgos físicos especiales que sugerían SS.
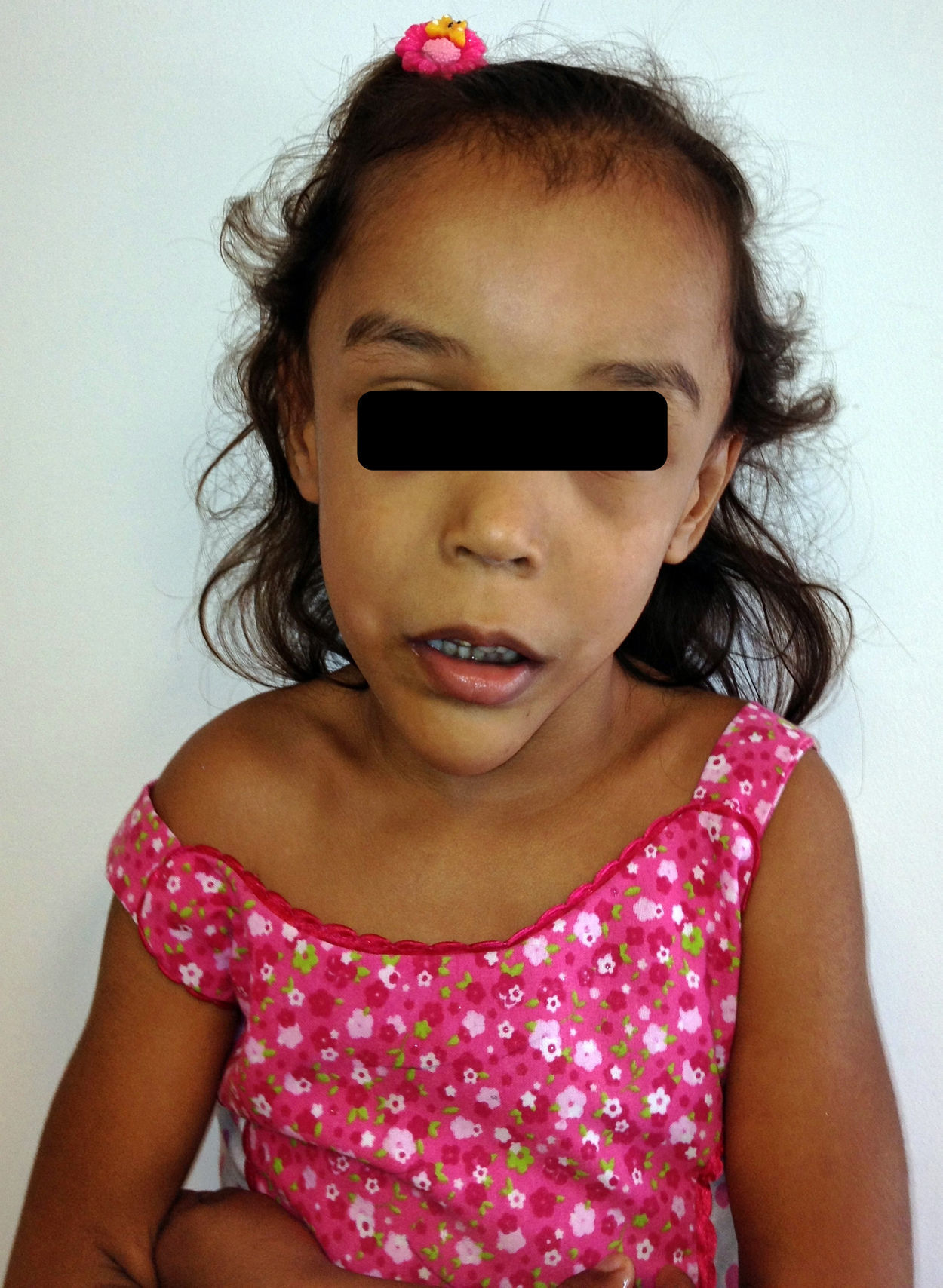
Caso clínicoPaciente de género femenino de 4 años de edad al momento de la evaluación, observándose al examen físico: talla de 106cm en percentil 75-90, peso de 17kg en percentil 50-75 DS 0,32 y perímetro cefálico 52cm en percentil 90, DS 1,88; cabello ralo, de pobre calidad, fascies triangulares, frente amplia, fisura palpebral alargada, filtrum largo, paladar ojival, mandíbula prominente, amelogénesis imperfecta; escápula alada y clinodactilia del quinto dedo en las manos, longitud de la mano 12,5 percentil 75-97, longitud del pie 17 percentil 75-97 (fig 1).

Como antecedentes se encontró: producto de unión entre padres no consanguíneos; al nacimiento madre de 20 años en su primera gestación, durante la cual no presentó enfermedades relevantes; 3 ecografías obstétricas que no revelaron anomalías congénitas; al nacimiento, edad gestacional de 39 semanas, parto vaginal con período expulsivo prolongado que requirió fractura de clavícula; recién nacida con peso de 3.600g, talla de 54cm y perímetro cefálico de 38cm; se sospechó asfixia perinatal severa que requirió hospitalización durante 25 días con presencia de un episodio convulsivo. La paciente, durante su infancia, presenta retardo marcado del desarrollo psicomotor y trastorno del lenguaje, pronunciando solo 10 palabras sin formación de frases completas, con pobre respuesta a terapia; bipedestación a los 3 años; actualmente no presenta control de esfínteres; cursa con reflujo gastroesofágico que requiere manejo farmacológico; presenta hiperactividad, no escolarizada por dificultad para el aprendizaje. Como estudios complementarios presenta: ecocardiograma, potenciales auditivos, TAC cerebral, ecografía renal, TSH y T4, todos estos con resultados en parámetros normales. Cariotipo bandas G con resolución de 650 bandas 46, XX.
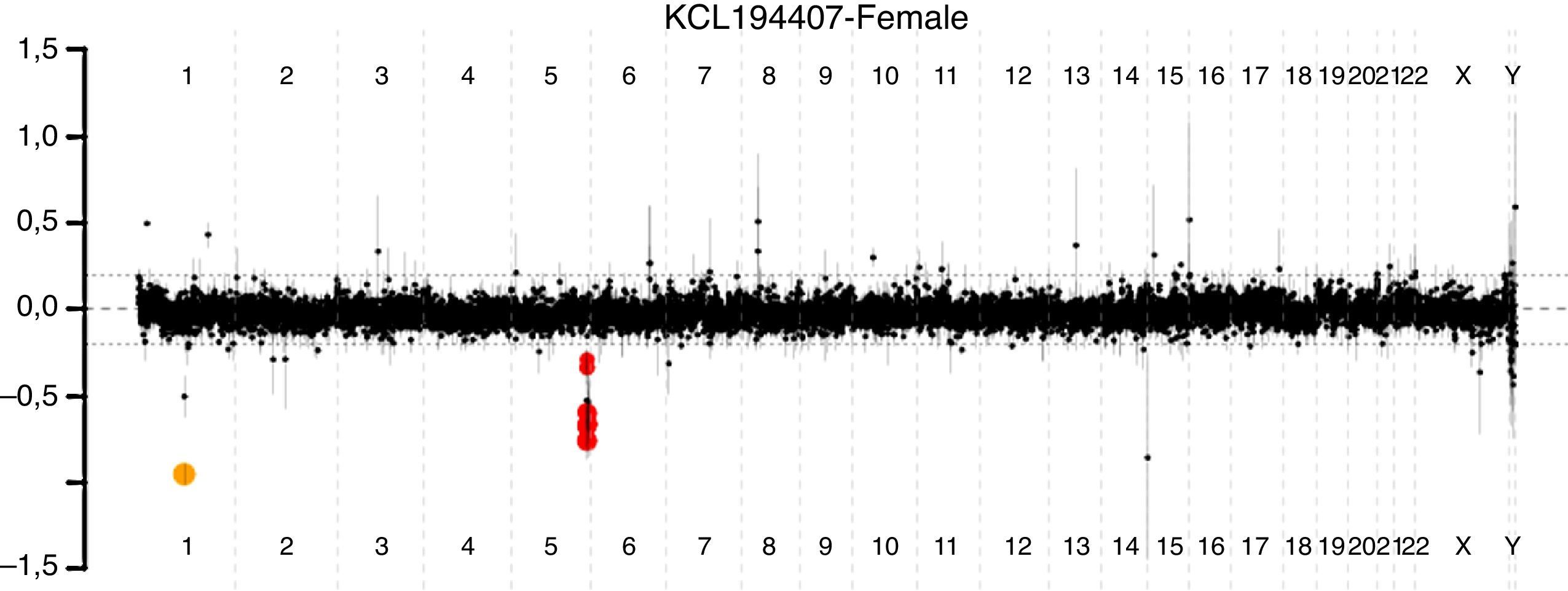
Se concluye que la sumatoria de hallazgos neurológicos y al examen físico no sería explicada por la probable asfixia perinatal, y que la etiología del fenotipo tiene un origen muy probablemente genético. Se estableció como primera posibilidad diagnóstica SS. Sin embargo, dado que los hallazgos no eran contundentes, y que podrían ser originados por un síndrome por microdeleción o microduplicación (incluyendo el SS), se solicita prueba de hibridación genómica comparativa por microarreglos, la cual concluye deleción 5q35.2 q35.3 de 2.082 MB; esta deleción abarca el gen NSD1 (fig. 2). Se confirma el diagnóstico de SS por deleción en la región 5q35, OMIM #1175505.

Análisis cromosómico por microarreglos de alta resolución de todo el material genético. Los puntos negros indican material genético en parámetros normales, sin exceso o déficit. En rojo se muestra pérdida de material genético del brazo largo del cromosoma 5, de 2.082 MB correspondiente a la región 5q35.2 q35.3 que incluye el gen NSD1.
La madre de la paciente firma consentimiento informado para tomar fotografías y utilizar los datos de la historia clínica.
DiscusiónEl SS fue descrito por primera vez en 1964 por el uruguayo Juan Sotos, quien describió 5 casos de niños con crecimiento excesivo y rápido, acromegalia, retraso del desarrollo psicomotor, paladar alto, mandíbula prominente y edad ósea avanzada6. El SS es una enfermedad rara, con una prevalencia de 7 en 100.000 nacimientos1, siendo uno de los síndromes más frecuentes de sobrecrecimiento, después de los síndromes de X frágil y Marfan.
El SS es causado en el 90% de los casos por haploinsuficiencia del gen NSD1 secundaria a mutaciones o deleciones, en el resto de los casos no se ha podido establecer la causa. Este gen tiene un locus 5q35, consta de 23 exones y codifica para una histona metiltransferasa, una proteína de 2.696 aminoácidos que se expresa en el cerebro, el músculo esquelético, el riñón, el bazo, el timo y el pulmón, y que actúa como un factor de transcripción intermediario cuya función biológica aún no ha sido establecida con exactitud, proponiéndose una relación con genes que promueven el crecimiento6,7.
En la población europea con SS las mutaciones puntuales intragénicas son predominantes, y se han encontrado entre un 40% a 80% de los pacientes, mientras que en la población asiática esta alteración se presenta solo en un 12% de los afectados, siendo más común las microdeleciones. En estudios de correlación fenotipo-genotipo se ha encontrado que los pacientes con microdeleciones tienen mayor severidad en el retraso mental, sobre crecimiento menos pronunciado y mayor frecuencia de anomalías cardiacas y genitourinarias que los que tienen mutaciones puntuales6,8.
El SS presenta un patrón de herencia autosómico dominante, lo que indica que un individuo afectado tiene un 50% de probabilidad de que su descendencia resulte con el síndrome. Aproximadamente un 95% de los casos no son heredados y las alteraciones génicas son de novo. En el caso reportado en este artículo, los padres de la paciente no presentaban fenotipo sugestivo de SS, lo que implica que se trató de una deleción de novo.
En 1994 Cole y Hughes publicaron un estudio de más de 500 individuos con alteraciones en el gen NSD1, en el cual sugerían 4 criterios de diagnóstico clínico como los más relevantes: apariencia facial característica, crecimiento excesivo y acelerado con talla, perímetro cefálico, manos y pies por encima del percentil 97, edad ósea avanzada y retraso en el desarrollo psicomotor.
Los hallazgos típicos faciales descritos incluyen frente amplia con línea de pelo frontotemporal elevada, fisuras palpebrales, enrojecimiento malar, paladar ojival o arqueado, hipertelorismo aparente, cara larga, mentón prominente, pabellones auriculares grandes, alteraciones odontológicas como erupción prematura de los dientes o amelogénesis imperfecta2. Los hallazgos faciales más relevantes de nuestra paciente incluían frente amplia, fisura palpebral alargada, paladar ojival, mandíbula prominente y amelogénesis imperfecta (fig. 3).

El SS pertenece a un grupo de síndromes de sobrecrecimiento que puede presentarse in útero o después del nacimiento. Alrededor del 90% de los pacientes con esta afección presenta talla y perímetro cefálico con percentil entre 90 y 97 o 2 desviaciones estándar por encima de la media; la macrocefalia está presente en todas las edades, y con frecuencia se encuentra que los pies y las manos son muy grandes en relación con su cuerpo. La talla tiende a normalizarse en la vida adulta, superando la media por pocos centímetros. La pubertad se inicia en límites normales, sin compromiso significativo de la edad de la menarquia en las mujeres2. Nuestra paciente presentaba perímetro de talla, perímetro cefálico, longitud de manos y pies en percentiles 75 a 97, por lo cual estas características fueron sugestivas para sospechar el diagnóstico de SS.
El déficit mental en los pacientes con SS varía entre leve a grave, y está presente en el 80-85% de los casos. Se manifiesta con dificultad para el procesamiento verbal, el razonamiento abstracto y la escritura; a su vez, es frecuente la presencia de retraso en el desarrollo de las habilidades motoras y dificultad en el desarrollo del lenguaje verbal. Alrededor del 50% exhibe convulsiones y el 30% presenta alteraciones conductuales y trastornos psiquiátricos, tales como aislamiento social, agresividad, depresión, ansiedad y psicosis9. En la paciente aquí presentada el retraso global del desarrollo podría ser explicado a partir de la probable asfixia perinatal sugerida por los médicos tratantes en el período neonatal; sin embargo, estos se sobreponen con los de muchos síndromes de origen genético, incluyendo el SS.
Los estudios de imagen mediante la resonancia magnética nuclear o la tomografía axial computarizada en pacientes con SS han evidenciado dilatación y alargamiento de los ventrículos cerebrales sin hipertensión craneal asociada. Además, se ha encontrado con relativa frecuencia hipoplasia o agenesia del cuerpo calloso, prominencia y dilatación de los cuernos occipitales, macrocisterna magna y atrofia del vermis6. La paciente de este caso presentó tomografía axial computarizada cerebral sin anormalidades.
Otros hallazgos fenotípicos que pueden encontrarse en el SS son anomalías cardiacas entre un 20% al 25% de los pacientes, tales como comunicación interauricular, comunicación interventricular, ductus arterioso persistente y estenosis pulmonar y tricuspídea. Estas alteraciones no fueron encontradas en nuestra paciente en el ecocardiograma. Al nacer, aproximadamente un 70% de los pacientes con SS presentan ictericia prolongada, hipotonía y disfagia; en menor porcentaje se evidencian anomalías renales con reflujo vesicouretral e infecciones urinarias recurrentes, otitis media de repetición, alteraciones en el desarrollo dental, anomalías oftalmológicas como estrabismo, cataratas y atrofia macular, infecciones respiratorias de repetición, escoliosis, reflujo gastroesofágico, hernias inguinales y alteraciones endocrinológicas como hipotiroidismo primario10; sin embargo, cabe aclarar que hasta ahora no se ha descrito ninguna anomalía endocrinológica que explique el sobrecrecimiento en este síndrome.
Otro hallazgo reportado en la literatura es el riesgo aumentado que presentan estos pacientes para el desarrollo de neoplasias. Se ha observado en afectados por SS neuroblastoma, teratoma sacrococcígeo, leucemia linfoblástica aguda y tumor de Wilms, entre otros; sin embargo, no se recomienda la realización de pruebas de cribado para estos tumores11. Las alteraciones adicionales a las clásicas que presentó la paciente descrita abarcaron amelogénesis imperfecta, clinodactilia y reflujo gastroesofágico.
El diagnóstico diferencial del SS debe hacerse con otros síndromes con sobrecrecimiento, entre ellos el más importante es el síndrome X frágil. La prevalencia de este síndrome es mayor que la reportada en el SS, es estimada en 1 en 5.000 hombres y 1 en 4.000 a 6.000 mujeres. Es la segunda causa de retraso mental con origen genético, después del síndrome de Down. Su fenotipo es más evidente en hombres que en mujeres, presentando talla elevada, macrocefalia, cara alargada, mentón prominente, frente alta, laxitud ligamentosa y macroorquidismo de aparición en la pubertad8.
Otros síndromes con sobrecrecimiento que deben descartarse son: Beckwith-Wiedemann, Weaver, Nevo, Bannayan Riley Ruvalcaba, Marshall-Smith, Perlman y Marfan, entre otros6.
El diagnóstico de SS se sospecha cuando los pacientes presentan hallazgos fenotípicos sugestivos y se confirma con las pruebas genéticas moleculares que evalúan el gen NSD1, como la secuenciación que detecta mutaciones puntuales, Multiplex ligation-dependent probe amplification y Fluorescent in situ hybridization, que detectan microdelecciones que involucran el locus del gen. La mayoría de las personas afectadas no tienen anormalidades en el cariotipo. Rara vez una anormalidad citogenética, como el hallazgo de una translocación 5q35 resulta en SS.
Discusión: En la paciente aquí reportada se realizó la prueba de hibridación genómica comparativa, esto debido a su retardo global del desarrollo junto con los hallazgos físicos especiales que sugerían SS, pero que en su totalidad no eran concluyentes; se solicitó una prueba que evaluara todo el genoma y permitiera detectar o descartar otros síndromes causados por microdeleción o microduplicación, incluyendo el mismo SS. Basados en la recomendación del Colegio Americano de Pediatría proponemos que los médicos en países de Latinoamérica soliciten hibridación genómica comparativa a pacientes con retraso global del desarrollo y hallazgos fenotípicos particulares en los que no haya una clara sospecha diagnóstica, dadas las posibilidades de enviar las muestras a laboratorios de referencia internacional en los países en los que no se ha implementado esta prueba molecular.
Conflicto de interesesEste trabajo cumple con los requisitos sobre consentimiento/asentimiento informado, comité de ética, financiación, estudios animales y sobre la ausencia de conflicto de intereses según corresponda.
