


El sarcoma granulocítico (SG) es una lesión poco frecuente asociada a síndromes mielodisplásicos, mieloproliferativos o leucemias, aunque puede ser el primer hallazgo en un paciente previamente sano. Presentamos un SG que comenzó como compresión medular, en un paciente sin patología hematológica previa. Las imágenes radiológicas demostraron una lesión lítica en L1 que precisó cirugía urgente. Fue preciso realizar inmunohistoquímica de la muestra para llegar al diagnóstico. El aspirado medular no mostró evidencia de patología hematológica, siendo el SG la primera manifestación. El paciente recibió posteriormente tratamiento con quimioterapia y radioterapia, falleciendo 20 meses después del diagnóstico de una sepsis Pseudomonas aeruginosa intratratamiento de una leucemia mieloblástica. En resumen, el SG primario es un tumor infrecuente de difícil diagnóstico. Es necesario tener un alto grado de sospecha y solicitar amplios estudios inmunohistoquímicos para un diagnóstico correcto. El tratamiento debe ser precoz, agresivo e individualizado, ya que tiene mal pronóstico.
Granulocytic sarcoma (GS) is an infrequent lesion associated with myelodysplastic or myeloproliferative disorders or leukemia, although it may be the first finding in an otherwise healthy patient. A case of GS is described that presented as spinal cord compression, in a patient with no underlying hematological disorder. Imaging studies disclosed a single lytic lesion in L1, which required emergency surgery. Immunohistochemical staining of the surgical biopsy sample was needed for diagnosis. Bone marrow aspirate was unremarkable. The patient received chemo-radiotherapy, dying 20 months after diagnosis of Pseudomonas aeruginosa sepsis during treatment of acute myelogenous leukemia. In short, primary GS is an infrequent and difficult to diagnose tumor. A high degree of suspicion, along with extensive immunohistochemical studies are necessary for diagnosis. Treatment should be prompt, aggressive and individualized, since the prognosis is very poor.
Presentamos el caso de un varón de 45 años que consultó por dolor lumbar.
Observación clínicaSe trata de un paciente varón de 45 años, natural de Marruecos, que llevaba 25 años viviendo en España. Consultó seis meses antes por un dolor intenso, tras un esfuerzo físico, en parrilla costal y fosa renal derecha. Fue diagnosticado de cólico renoureteral y recibió tratamiento analgésico y antiinflamatorio, pudiendo reanudar su trabajo a los dos meses. Los cuatro meses siguientes, persistió una discreta molestia no incapacitante en la zona. Dos semanas antes de acudir al Servicio de Urgencias, presentó nuevamente, tras un esfuerzo físico, dolor brusco en dicha localización, precisando derivados opioides por la ausencia de mejoría con analgésicos habituales. Asociaba pérdida de peso no cuantificada los últimos meses, sin otra clínica. El paciente presentaba un hábito asténico, sin otros hallazgos.
ResultadosSu hemograma mostraba recuentos y fórmula leucocitaria en rangos de normalidad y no se detectaron alteraciones bioquímicas sanguíneas incluyendo proteínas totales, TSH, B2-microglobulina, inmunoglobulinas, espectro electroforético, PSA, CA19.9, CEA y la orina. El frotis de sangre periférica era normal, al igual que: Mantoux, rosa de Bengala, triptasa sérica, la ecografía cervical y la gastroscopia.
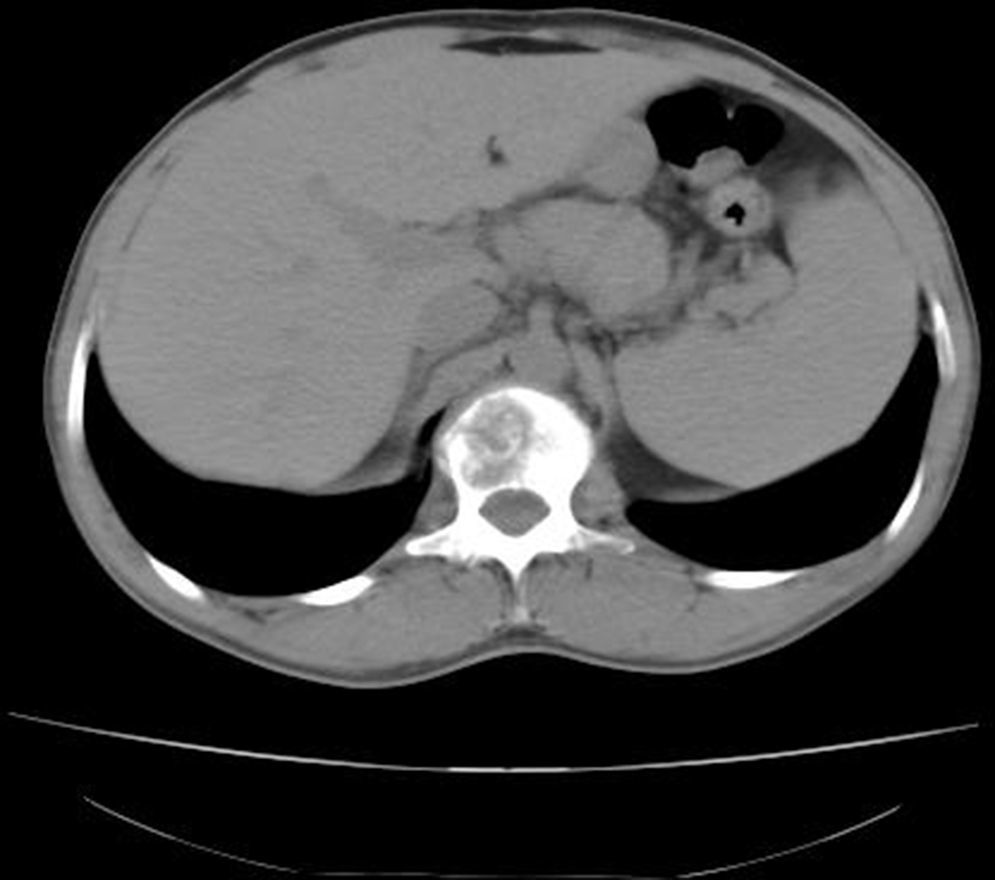
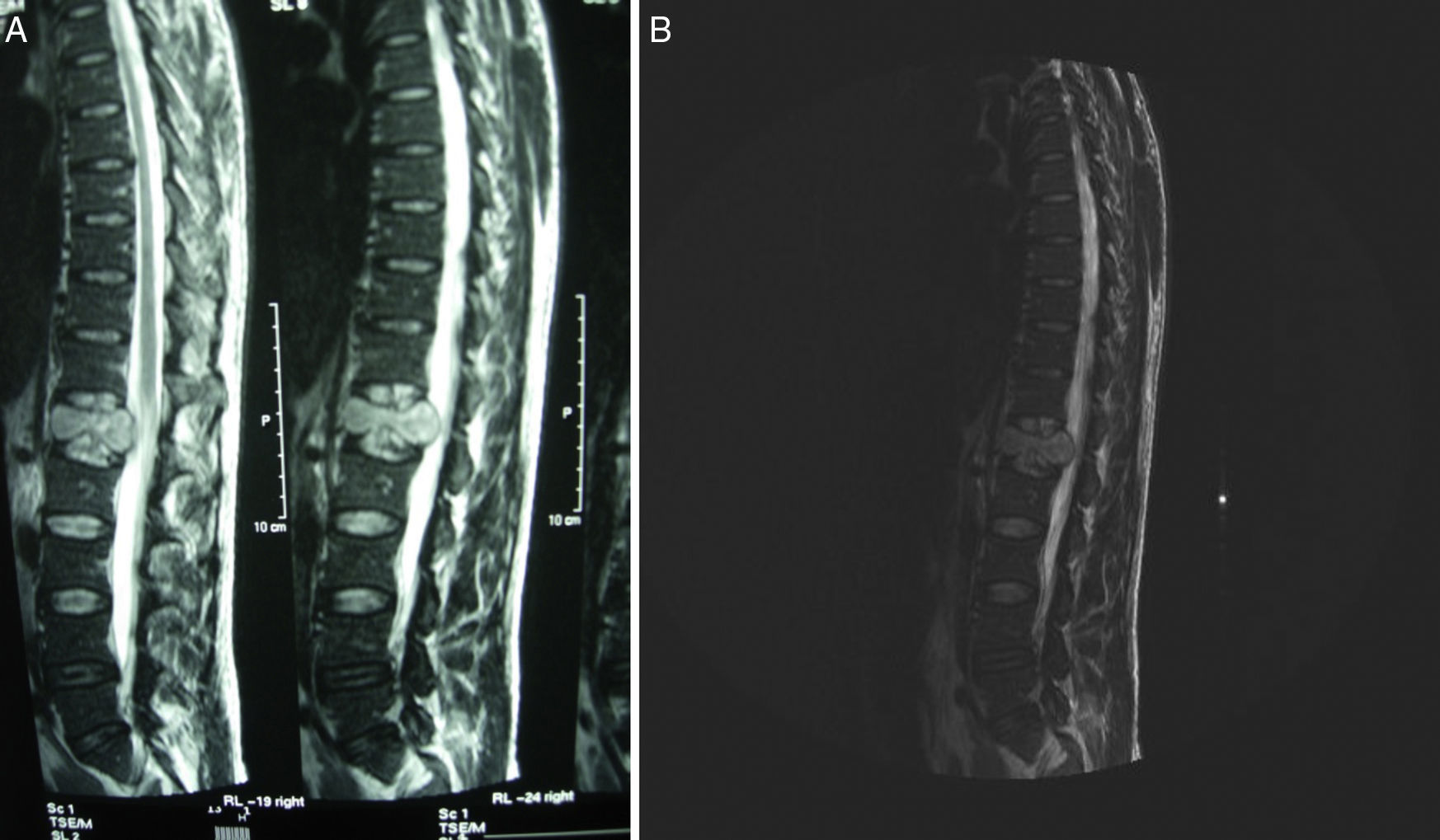
La tomografía axial computarizada (TAC) toraco-abdomino-pélvica con contraste y la resonancia magnética (RM) lumbar mostraron una lesión lítica amplia en el cuerpo vertebral L1 con afectación del pedículo derecho y un componente de partes blandas que excedía el contorno del propio cuerpo vertebral protruyendo a la porción anterior del conducto raquídeo con leve compresión del mismo (figs. 1 y 2). La gammagrafía ósea confirmó la lesión vertebral como única. Se realizó una punción aspiración con aguja fina guiada por tomografía axial computarizada (muestra no representativa). Ante estos hallazgos, se decidió realizar corporectomía de L1 urgente con fijación anterior e injerto óseo.

 con marcado abombamiento del muro posterior.")
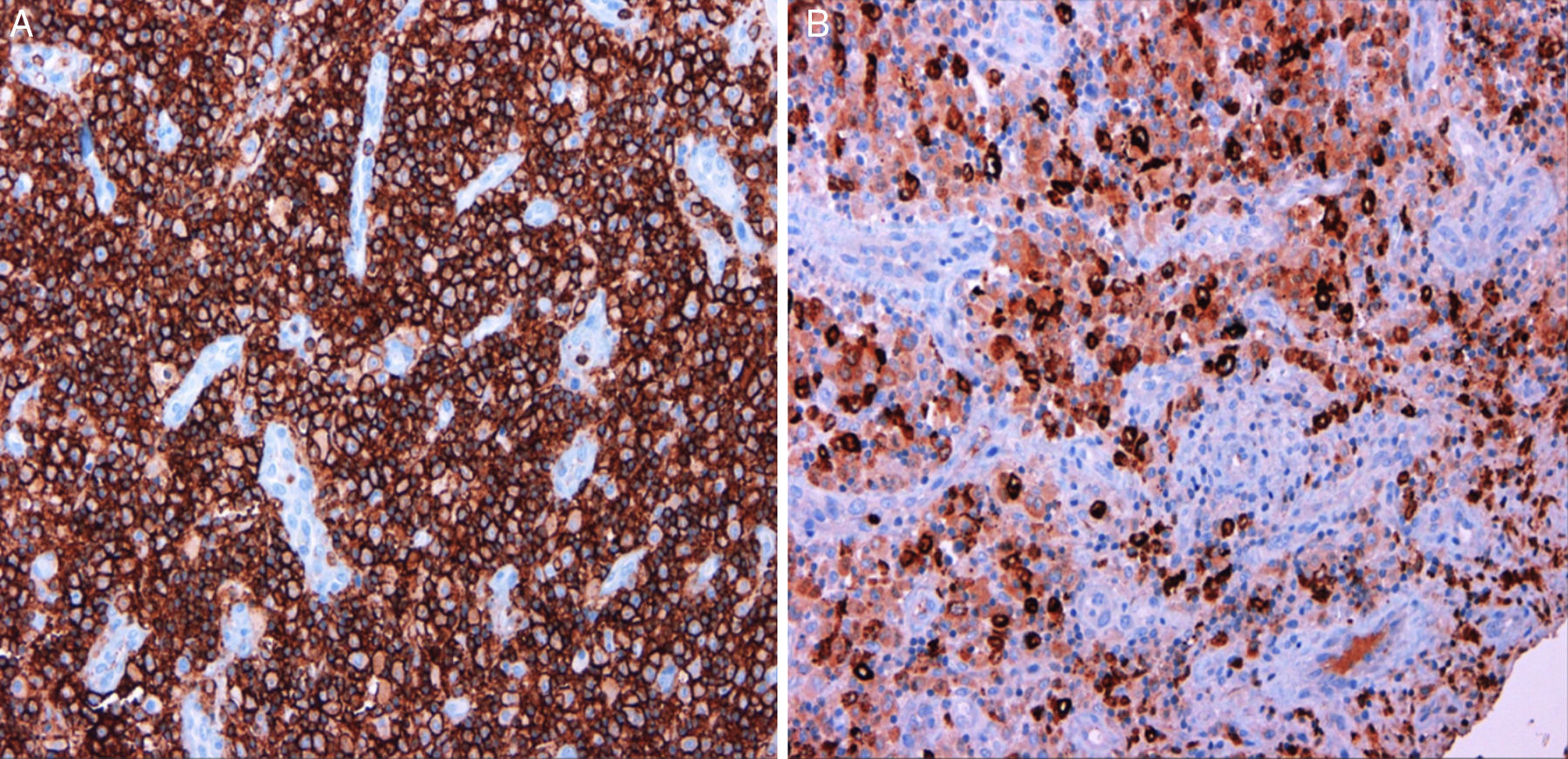
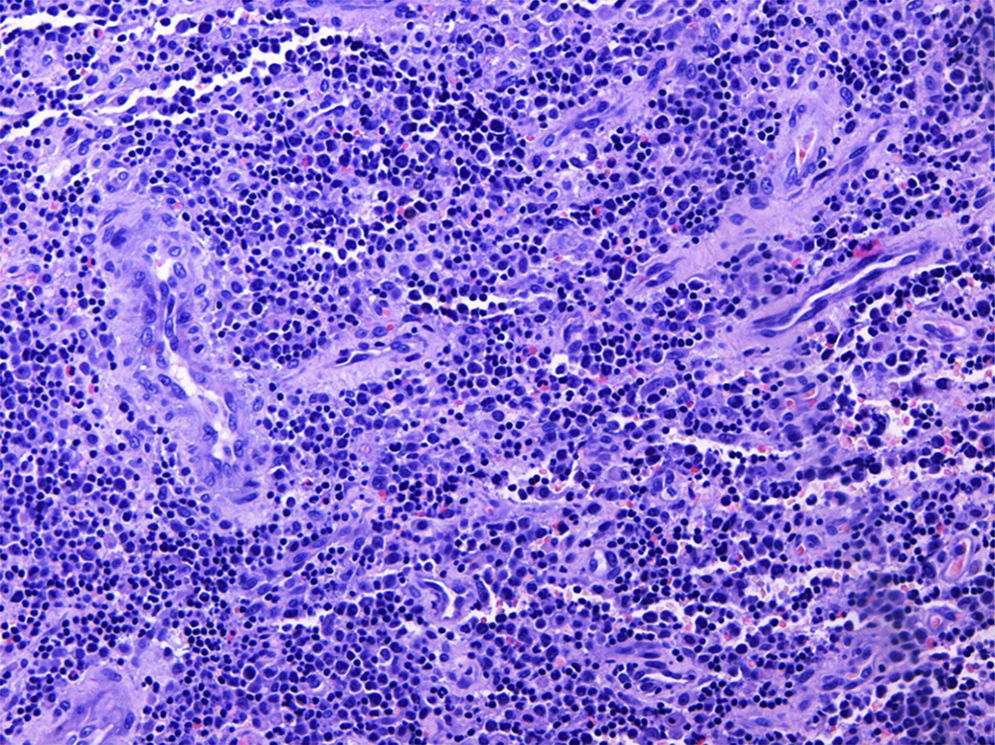
La anatomía patológica reveló una lesión compatible con un sarcoma granulocítico, constituida mayoritariamente por células hematopoyéticas de aspecto inmaduro con estudio inmunohistoquímico positivo para CD45, CD34, CD43, mieloperoxidasa (MPO), CD15 y CD68, negativo para CD1-a, CD3, CD20, CD30, S100 y antígeno epitelial de membrana, y con un índice de replicación bajo (Ki67 30%) (fig. 3). El tejido esponjoso y fibroso periostal circundante presentaba hiperplasia de la médula hematopoyética con algunas células inmaduras (fig. 4). La biopsia de médula ósea no mostraba alteraciones citológicas, inmunofenotípicas o citogenéticas, por lo que la lesión se consideró un sarcoma granulocítico (SG) primario.
. El estudio inmunohistoquímico muestra una intensa positividad difusa de las células para CD43 (imagen izquierda), que también es positiva para mieloperoxidasa (imagen derecha).")
. Tejido fibroso periostal con denso infiltrado celular polimorfo, donde se reconocen linfocitos con aislados eosinófilos y células de mayor tamaño, de aspecto inmaduro y de aspecto blástico.")
Se inició quimioterapia sistémica con idarrubicina y citarabina (IA 3+7), y se asociaron seis dosis de quimioprofilaxis triple intratecal. Posteriormente, recibió radioterapia sobre D12, L1 y L2 (dosis total 30Gy). Se mantuvieron controles clínicos, radiológicos y analíticos periódicos, sin intensificación del tratamiento dada la neutropenia severa que presentaba el paciente. Ingresó por leucemia mieloide aguda (LMA) 13 meses después del último ciclo de quimioterapia, presentando cambios mielodisplásicos y presencia en el análisis citogenético de trisomía del cromosoma 8 y deleción del brazo largo del cromosoma 7. Se inició entonces tratamiento quimioterápico según esquema FLAG-Ida (fludarabina, citarabina, idarrubicina y filgastrim). En el día +24, el paciente fallece tras presentar shock séptico secundario a bacteriemia por Pseudomona aeruginosa. El fallecimiento sucedió a los 20 meses del diagnóstico, dos años después del inicio de la sintomatología.
DiscusiónEl SG o sarcoma mieloide se define como un tumor extramedular constituido por células hematológicas mieloides. Burns1 lo describió por primera vez en 1811 en un paciente con una lesión retroorbitaria de coloración verdosa. En 1853 King2 lo denominó «cloroma», del griego chloros, verde. Este aspecto característico es debido a la presencia de MPO en los mieloblastos3.
Se trata de una neoplasia infrecuente cuya presentación clínica más común es su aparición en pacientes con patología hematológica previa, ya sea una LMA, síndrome mielodisplásico o mieloproliferativo. Es menos habitual que suponga la primera manifestación de estas enfermedades y excepcional que se manifieste como una lesión aislada o primaria sin evidencia en el estudio de médula ósea de hemopatía subyacente. En 1981 Neiman et al. publicaron una serie de 61 casos de SG de los cuales un 36% se presentaban sin trastorno mieloproliferativo asociado4. Pileri et al. publicaron en el 2007 la serie más larga hasta el momento, con 92 casos, siendo el 27% SG primarios5. En general, la incidencia estimada en adultos de un SG primario es de tan solo 2/1.000.0004.
El SG puede afectar cualquier parte del cuerpo, de forma solitaria o múltiple, pero se localiza más frecuentemente en hueso, piel y ganglios linfáticos. Su clínica es poco específica, debida principalmente al efecto de masa y disfunción del órgano afecto.
Su diagnóstico de certeza precisa estudios histológicos, ya que no tiene características radiológicas o marcadores bioquímicos que permitan distinguirlo de otras neoplasias. Evidentemente, el contexto clínico puede ser determinante, ya que la coexistencia de una hemopatía mieloide no controlada puede hacer hasta cierto punto innecesaria la biopsia de la tumoración.
Aquellos pacientes cuya única clínica es un dolor circunscrito subagudo o crónico, que finalmente resulta ser la manifestación de un tumor puesto en evidencia por las imágenes radiológicas, en una localización difícilmente accesible y sin datos adicionales en otras pruebas complementarias suponen en general un reto diagnóstico. El SG, por su infrecuencia y desconocimiento, no suele considerarse como una posibilidad, y menos si en el estudio solicitado se incluye una biopsia de médula ósea anodina.
Este es el caso de la mayoría de los SG localizados en la columna vertebral, que suponen el 13% del total de los primarios de acuerdo a la serie de Yamauchi et al.6 y no presentan por lo general clínica neurológica al comienzo. Suelen afectar un segmento único, más frecuentemente en vértebras dorsales, seguidas de las lumbares, aunque se describen casos con afectación más extensa a lo largo de la columna7,8. Lógicamente, suelen pasar por dorsolumbalgias mecánicas o cólicos renoureterales en sus primeras consultas. En el paciente que presentamos, la cirugía se retrasó cuatro semanas debido a la falta de especificidad del dolor, siendo necesaria una intervención urgente al observar compresión medular por el tumor en la RM solicitada. El resultado de la anatomía patológica se demoró otras cuatro semanas debido a la complejidad de su estudio.
Aun disponiendo de muestras de tejido tumoral, el diagnóstico de un SG puede resultar complejo y requerir la combinación de diversos estudios para evitar su confusión con otras neoplasias. Las series clásicas comunicaban errores en el diagnóstico de hasta un 75% de los casos9. En estudios más recientes este porcentaje se situaba aún en torno al 25-47%6,10, en la gran mayoría de los casos debido a la dificultad para diferenciar su morfología de la de los linfomas no Hodgkin de células B grandes. Ambas entidades muestran una celularidad patológica similar, de gran tamaño, citoplasma amplio y con tinciones básicas indistinguibles. Así la MPO, uno de los marcadores mieloides más característicos, resulta siempre negativa en las células linfoides pero puede serlo igualmente en los mieloblastos menos diferenciados11,12. Actualmente para lograr un diagnóstico de certeza deben por ello combinarse datos obtenidos de una inmunohistoquímica más amplia con estudios de hibridación fluorescente in situ (FISH) y análisis moleculares. La inmunocitoquímica es el método más práctico para establecer el diagnóstico del SG, y por lo general más sencillo que la citometría de flujo que precisa células en suspensión. El marcador expresado con más frecuencia es el CD68/KP1, también suelen presentar positividad para MPO, CD117, CD99, CD68/PG-M1, lisozima, CD34, deoxinucleotidil transferasa terminal (TdT), CD56, CD61, CD30, glucoforina A y CD413. Las expresiones antigénicas aberrantes son poco frecuentes y la combinación de estos antígenos permiten realizar un diagnóstico diferencial con linfomas no Hodgkin y con neoplasias no hematológicas como neuroblastomas, rabdomiosarcomas, sarcomas de Ewing, histiocitomas, timomas, carcinomas o hematopoyesis extramedular6,9,10.
En el estudio realizado por Pileri et al. en pacientes con SG primario y asociado a otra hemopatía, un 54% presentaban alteraciones citogenéticas, siendo las más frecuentes la monosomía del cromosoma 7 y la trisomía del 85.
En cuanto al tratamiento de los SG, no existe consenso, ni en los primarios ni cuando se asocian a otra hemopatía. Esto se debe a la falta de estudios prospectivos aleatorizados por su baja incidencia. Tsimberidou et al. documentaron que los pacientes tratados con regímenes de quimioterapia con citarabina, con radioterapia o sin esta, conseguían la remisión completa en un 65% de los casos14. Es necesario estudiar cada caso individualmente y plantear un tratamiento según distintos aspectos como la edad, comorbilidades o grado de afectación. Por lo general, los SG primarios son tratados inicialmente como una LMA, con regímenes de quimioterapia para la inducción a la remisión seguidos de consolidación. El tratamiento quirúrgico y la radioterapia suele indicarse cuando la masa es grande o como en nuestro caso si produce compresión medular. Tampoco está claro el papel del trasplante de médula ósea, tanto autólogo como alogénico, aunque se asocia a tasas de supervivencia global ligeramente más altas en la literatura10,12,15,16. Aunque la mayoría de los SG son radiosensibles, el tratamiento único con radioterapia no resulta beneficioso, y se emplea de forma localizada para lesiones óseas muy dolorosas o compresivas17.
El comportamiento clínico y la respuesta al tratamiento del SG no parecen estar influenciados por la edad, el sexo, la localización anatómica, el hecho de ser tumor primario o asociado a otras hemopatías, los hallazgos histológicos, inmunohistoquímicos o citogenéticos. En algunos casos, no obstante, el tratamiento precoz puede mejorar el pronóstico y disminuir la probabilidad de que evolucione a una enfermedad sistémica5,8,18.
En conclusión, el SG es un tumor infrecuente y de difícil diagnóstico a considerar en el diagnóstico diferencial de la compresión medular por tumores. Su aparición como primera manifestación de una enfermedad hematológica resulta todavía más infrecuente, por lo que es importante tener un alto grado de sospecha y solicitar amplios estudios inmunohistoquímicos para un diagnóstico correcto. Se recomienda iniciar un tratamiento sistémico precoz, agresivo e individualizado, ya que el SG primario se considera una enfermedad de mal pronóstico, similar al de una LMA14,15.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
