



La enfermedad de Camurati-Engelmann es un trastorno autosómico dominante, de baja incidencia, y sin predilección por sexo o raza. Su diagnóstico se basa en hallazgos clínicos y radiológicos. La hiperostosis diafisometafisaria afecta principalmente a los huesos largos y de forma secundaria a huesos planos como los de la bóveda craneal. Alteraciones hematológicas como anemia, leucocitopenia y esplenomegalia pueden presentarse asociadas con la enfermedad. No existe un tratamiento específico de la enfermedad. Se han reportado resultados variables con el uso de esteroides y resultados pobres con antirresortivos óseos. Reportamos un caso de un paciente de sexo masculino con hallazgos radiológicos y clínicos sugestivos de dicha enfermedad, con una fractura patológica subtrocantérica como complicación. Se describe la aproximación diagnóstica junto con los hallazgos radiológicos, clínicos e histopatológicos de confirmación, y se discute el manejo de este caso particular de enfermedad.
Camurati-Engelmann is an uncommon autosomal dominant disease, with no predilection for sex or race. Diagnosis is based on the clinical and radiological findings. The progressive diaphyseal and metaphyseal hyperostosis mainly affects the long bones and then the flat bones, such as those of the cranial vault. It is also associated with blood disorders including, anemia, leukopenia, and splenomegaly. There is no specific treatment for this disease, but several miscellaneous results with treatments using steroids, as well as with bisphosphonates have been described. We report a case of a man with radiological and clinical findings suggestive of this disease, who presented with a subtrochanteric fracture in one of the sites of expression of this bone disease. We describe the diagnostic approach that led to this disease suspicion, and further clinical, radiological, and histopathological findings, in order to present a discussion on its management.
La enfermedad de Camurati-Engelmann (ECE), también llamada displasia diafisaria progresiva, se ha identificado como un trastorno autosómico dominante, descrito inicialmente por Cockayne en 19201,2; en 1922, Camurati propuso que el trastorno era hereditario3. En 1929, siete años más tarde, Engelmann describió clínicamente la forma grave de la enfermedad4.
Es una enfermerdad rara, con una incidencia de 1 caso por millón de habitantes, y no se ha observado diferencias significativas en la frecuencia por sexo o raza5. El diagnóstico se basa esencialmente en el análisis de los datos de la anamnesis, hallazgos semiológicos y radiológicos.
Por lo general, se manifiesta durante la infancia, con dolor en las extremidades inferiores, cojera (típicamente el paciente aumenta la base de sustentación), signo de Trendelemburg tardío, atrofia muscular, fatiga, debilidad y disminución del trofismo del tejido celular subcutáneo en las extremidades, que puede parecer una distrofia muscular como la enfermedad de Duchene.
En un alto porcentaje de pacientes, se observan un hábito corporal marfanoide y estatura en percentiles altos para la edad, macrocefalia con frente prominente y proptosis, y en algunos casos se presenta parálisis de pares craneales o aumentos de presión intracraneana, que pueden aparecer inadvertidamente. Estos hallazgos se deben al sobrecre cimiento óseo en los orificios craneanos, que puede producir compresión de nervios periféricos, especialmente los nervios craneales VI, VII, VIII, y de estructuras del sistema nervioso central, como el bulbo raquídeo y las amígdalas cerebelosas, por su vecindad con el foramen magno.
Es frecuente encontrar manifestaciones hormonales como pubertad tardía con hipogonadismo secundario y crecimiento óseo prolongado. En cuanto al sistema muscular, se puede encontrar un síndrome crónico de desacondicionamiento, con contracturas en caderas y rodillas en flexión, especialmente, además de dolores musculares generalizados e inespecíficos. El principal hallazgo radiológico de la enfermedad de Camurati-Engelmann es la hiperostosis de la diáfisis de los principales huesos largos, producida por la proliferación de tejido óseo en la superficie perióstica y endoostio. La maduración de la neoformación ósea secundaria acentúa el crecimiento cortical.
Los huesos largos más involucrados son las tibias y los fémures, con menor frecuencia los huesos largos de la extremidad superior y los tubulares cortos. La hiperostosis, por lo general, tiene inicio en la diáfisis y al crecer involucra las metáfisis y por lo general asume un patrón simétrico, y característicamente no afecta a las epífisis. Puede afectar a otros huesos como los del oído medio, produciendo otosclerosis, y también puede implicar el cráneo y el esqueleto axial, las clavículas, los omóplatos y la pelvis.
Se puede encontrar manifestaciones sistémicas hematológicas tales como anemia, leucocitopenia y hepatoesplenomegalia. En los casos graves, hay una afección generalizada y se puede observar áreas de osteopenia, e incluso se puede observar en la gammagrafía áreas de aumento de captación de radionucleótidos cuando hay lesiones activas y precoces.
La imagen radiológica típica en huesos largos se observa en el 94% de los pacientes con mutaciones identificables, un 54% además tiene manifestaciones craneales y un 63%, en la pelvis. En la resonancia magnética se puede observar trastornos de la médula ósea, y el microscopio electrónico puede revelar miopatía y cambios vasculares, como atrofia de fibras musculares sin cambios degenerativos.
En estudios histológicos se encuentran conglomerados de neoformación ósea desorganizados, con maduración centrípeta, engrosamiento de la cortical y estrechamiento de la cavidad medular. Los biomarcadores de recambio óseo pueden elevarse, incluida la fosfatasa alcalina sérica. También son frecuentes la hipocalcemia y leve hipocalciuria, anemia leve, leucocitopenia y elevación de la velocidad de eritrosedimentación globular (VSG).
En el año 2000, se descubrió que la mutación del gen que codifica el factor de crecimiento transformador β1 (TGFβ1) en 19q13 y alteraciones en los genes que regulan la importante homeostasis del esqueleto (TGFB1 y TNFSF11), la codificación de TGFβ1 y ligando RANK (RANKL). Se encontró asociación con este gen en un 94% de los pacientes con signos radiológicos de ECE.
En cuanto al tratamiento farmacológico, los glucocorticoides son agentes antiinflamatorios e inmunosupresores, que son la piedra angular del tratamiento. En el hueso, disminuyen la densidad ósea, aumentando la tasa de apoptosis de los osteoblastos y osteocitos, mientras que suprime la proliferación de osteoblastos y la diferenciación y la síntesis de la matriz ósea. Tratamientos de prednisolona hasta 1mg/kg/día son los más utilizados por ciclos indefinidamente, teniendo en cuenta los efectos adversos de los esteroides a mediano y largo plazo. No se ha encontrado buenos resultados con el tratamiento con bisfosfonatos como pamidronato y etidronato, incluso se han presentado efectos adversos como, por ejemplo, elevación de fosfatasa alcalina6. Otros fármacos utilizados incluyen los antiinflamatorios no esteroideos (AINE), como el ácido acetilsalicílico, que aunque pueden aliviar el dolor, no son eficaces para mejorar los cambios óseos ya establecidos.
El tratamiento quirúrgico es una alternativa al farmacológico y para tratar complicaciones óseas. Se ha planteado realizar el fresado del canal para aliviar el dolor y evitar el sobrecrecimiento del canal medular y los defectos de modelado, además del tratamiento con osteosíntesis de las fracturas asociadas.
El diagnóstico diferencial de esta enfermedad incluye afecciones como la displasia fibrosa, la fluorosis, la intoxicación por metales pesados, la osteodistrofia renal, la hipovitaminosis A, la sífilis, la osteomielitis y la osteopetrosis.
- •
También hay otras enfermedades enmarcadas en un cuadro sindrómico, que pueden ser similares y ocasionar controversia en el momento de diagnosticar una ECE:
- •
Enfermedad de Ribbing: causada por mutaciones en TFGβ1, produce manifestaciones clínicas similares7.
- •
Displasia craneodiafisaria: engrosamiento de los huesos del esqueleto axial que incluyen el cráneo y producen hipertelorismo ocular, proptosis y prominencia frontal y afección diafisaria de huesos largos.
- •
Síndrome de Kenny-Caffey tipo 2: síndrome en el que se encuentra enanismo, acompañado de engrosamiento cortical de los huesos largos y retardo en el cierre fontanelar.
- •
Anomalías craneofaciales, hipocalcemia e hipoparatiroidismo.
La terapia génica se considera como el futuro del tratamiento de esta enfermedad, y ya se han publicado estudios in vitro en animales con mutación del gen TGF-1.
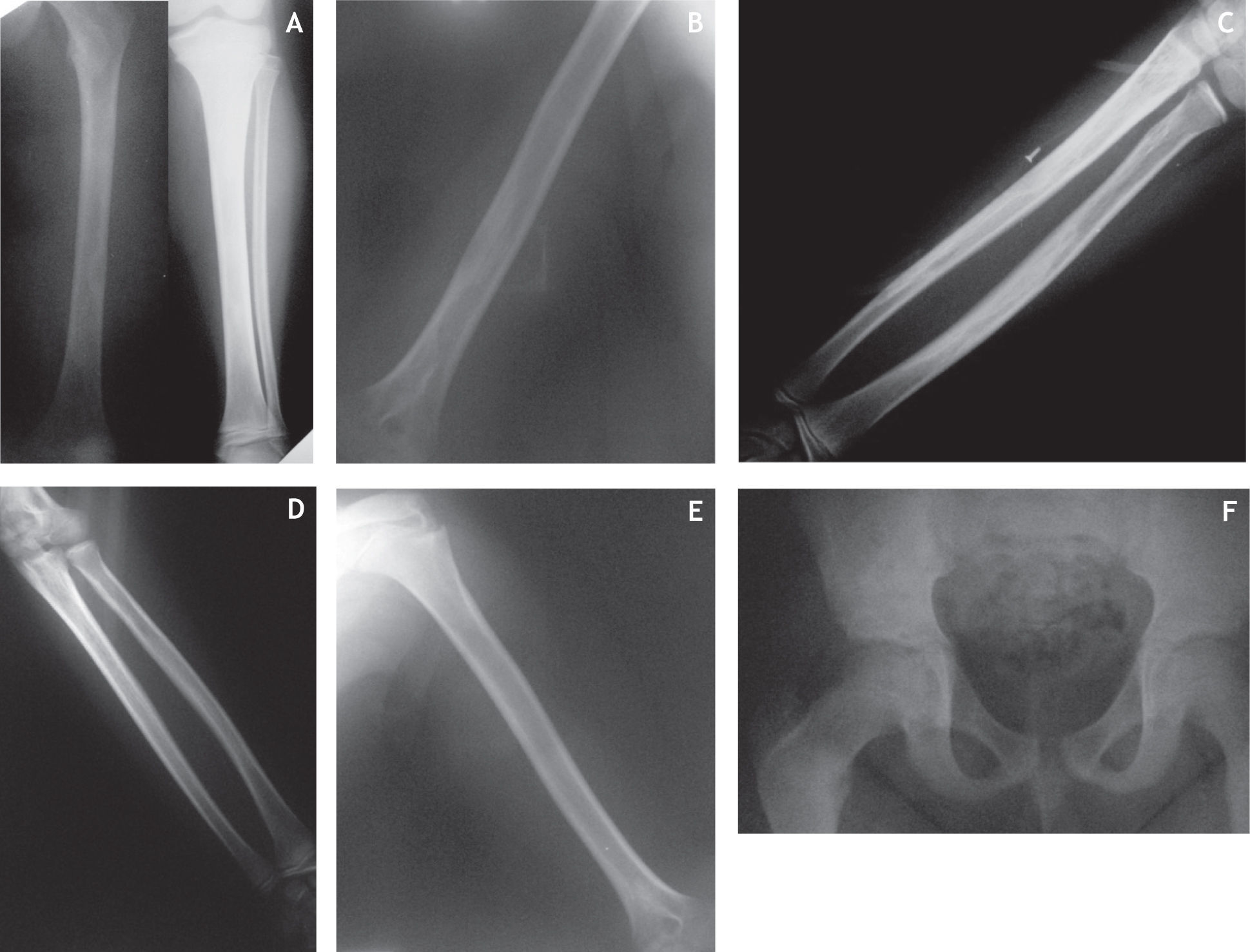
Caso clínicoPaciente de 11 años que consultó por dolor en las extremidades superiores e inferiores sin relación con actividad física y que se presenta en ocasiones por la noche; es inespecífico, sin irradiación ni remisión de los síntomas con analgésicos y AINE, y su evolución aproximada es de 1 año. Estudios preliminares mostraban lesiones diafisarias en extremidades superiores e inferiores, evidenciadas en imágenes radiográficas de las extremidades en mención con lesiones radioopacas que comprometían las diáfisis de huesos largos, húmeros, antebrazos y tibias con mayor compromiso en porciones proximales de ambos fémures, y ocluyendo el canal medular de los huesos tubulares largos, como se muestra en la figura 1.

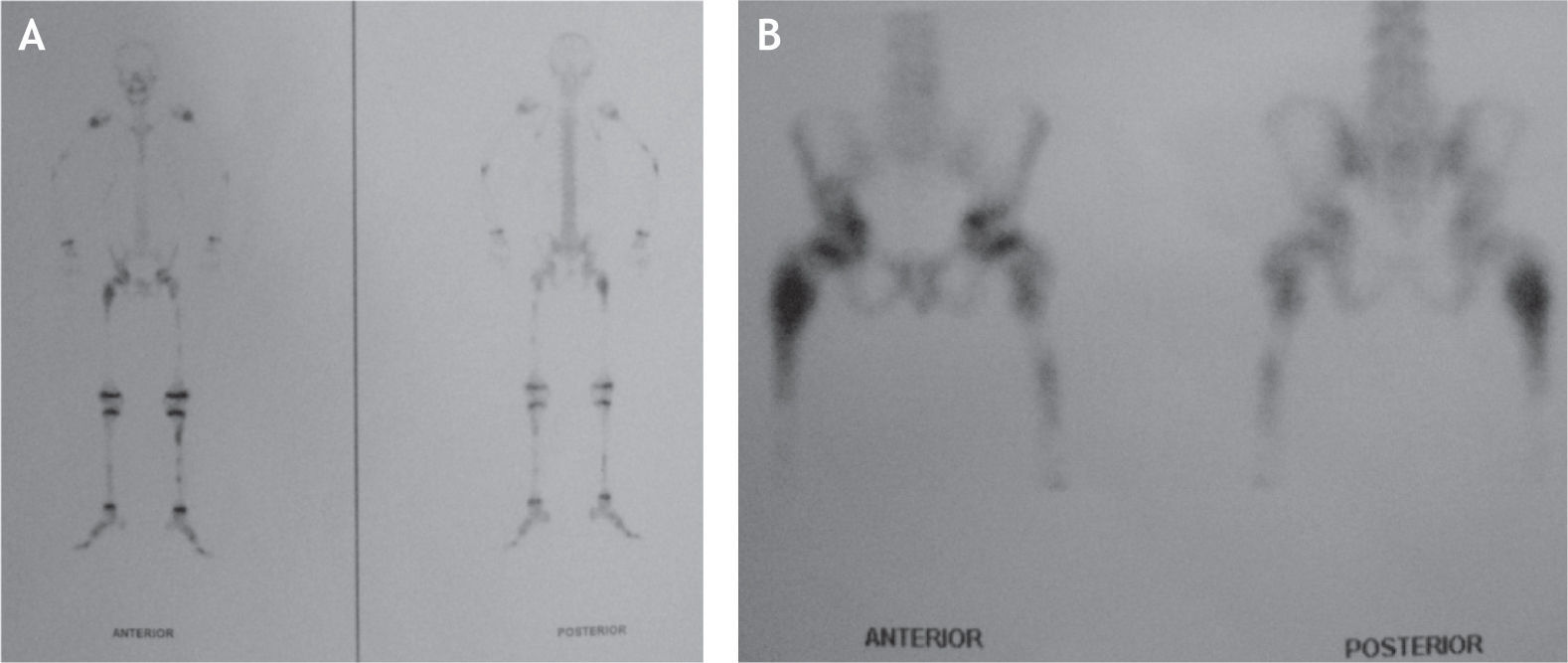
Los anteriores estudios complementados con gammagrafía ósea de tres fases mostraban hallazgos compatibles con enfermedad poliostótica multicéntrica de huesos largos, a descartar por los hallazgos de neoplasia hematológica como leucemia; no se observaba distribución anormal en fase tisular y vascular, pero se apreciaban en la fase ósea focos de hipercaptación en los fémures, con mayor intensidad en sus tercios proximales, tibias y húmeros, sin afección en huesos cortos y/o planos (fig. 2).

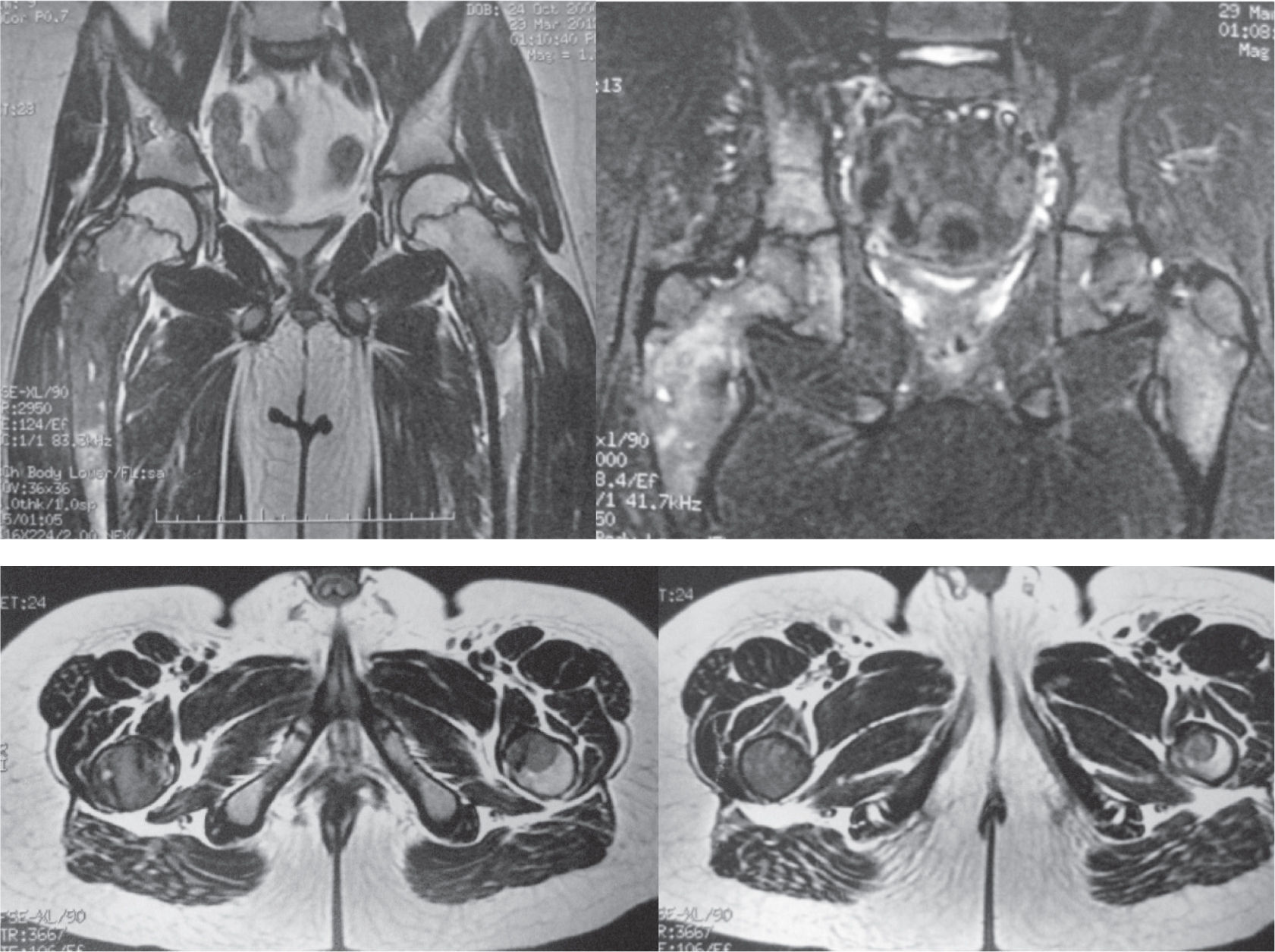
Estudios por resonancia magnética practicados en controles posteriores muestran en ambas caderas lesiones que comprometen e infiltran el canal medular, en regiones intertrocantéricas y subtrocantéricas de los fémures, con signos de edema de médula ósea sin afección extracortical (fig. 3).

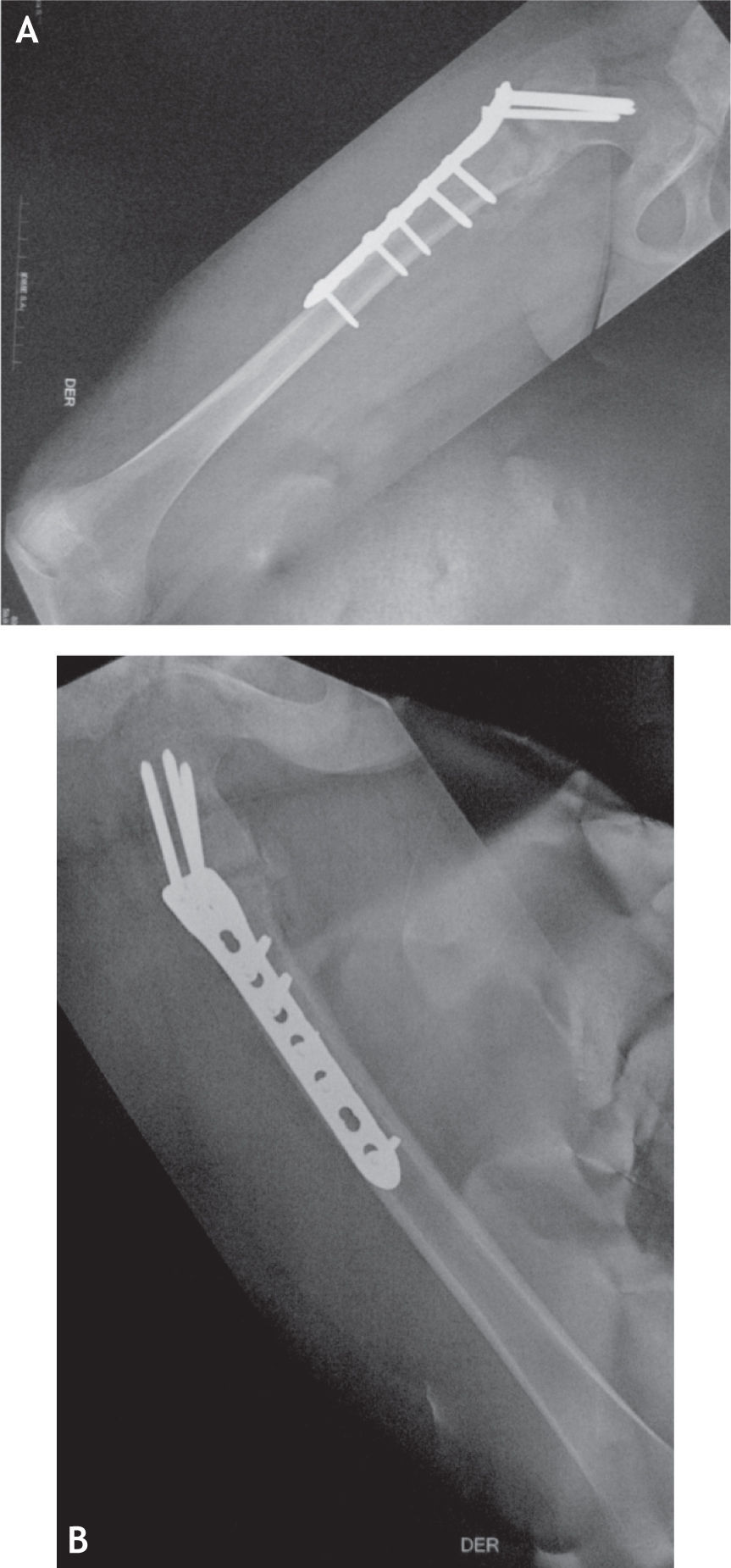
El paciente persistía con dolores en ambas extremidades, con la consecuente disminución de la actividad física. Posteriormente tuvo una caída al tropezar mientras descendía por escaleras, y sufrió fractura subtrocantérica en fémur derecho (figs. 4 y 5) no expuesta, que requirió reducción abierta más fijación con material de osteosíntesis, en este caso LCP para fémur proximal; a la vez se tomó biopsia de canal medular y se fresó.


El estudio de histopatología informa de proliferación fibroósea benigna con trabéculas óseas inmaduras confluentes, estroma intertrabecular fibroconectivo denso y laxo y evidencia de medular estrecha, osteoclastos peritrabeculares para diagnóstico de displasia esclerosante ósea, favoreciendo diplasia diafisaria progresiva congruente con enfermedad de Camurati-Engelman.
El paciente recuperó la función articular paulatinamente, con movilidad completa de la cadera y de rodilla, y el control radiográfico muestra consolidación en progreso.
DiscusiónLa enfermedad de Camurati-Engelmann, dada su rara incidencia, puede pasar inadvertida y aparecer en el marco de síntomas inespecíficos, hallazgos radiológicos incidentales o al documentar fracturas patológicas que obligan al médico tratante a solicitar series óseas.
Más aún, se encuentra gran dificultad en delimitar con los hallazgos semiológicos y radiológicos de dicha enfermedad, teniendo en cuenta los diagnósticos diferenciales que son muy similares en su semiología, y es así como el examen histopatológico se considera una herramienta fundamental para poder definir y concretar la enfermedad. Es discutible pensar en criterios clínicos diagnósticos porque la ECE puede acompañarse de manifestaciones sistémicas o afecciones en sistemas diferentes del osteomuscular.
En el caso clínico presentado, se realizó un análisis tanto clínico como radiológico de la enfermedad en la búsqueda de delimitar claramente los hallazgos semiológicos clínicos y radiológicos, teniendo en cuenta que las características antropométricas o el hábito del paciente no son el habitual longilíneo que se relaciona con dicha enfermedad.
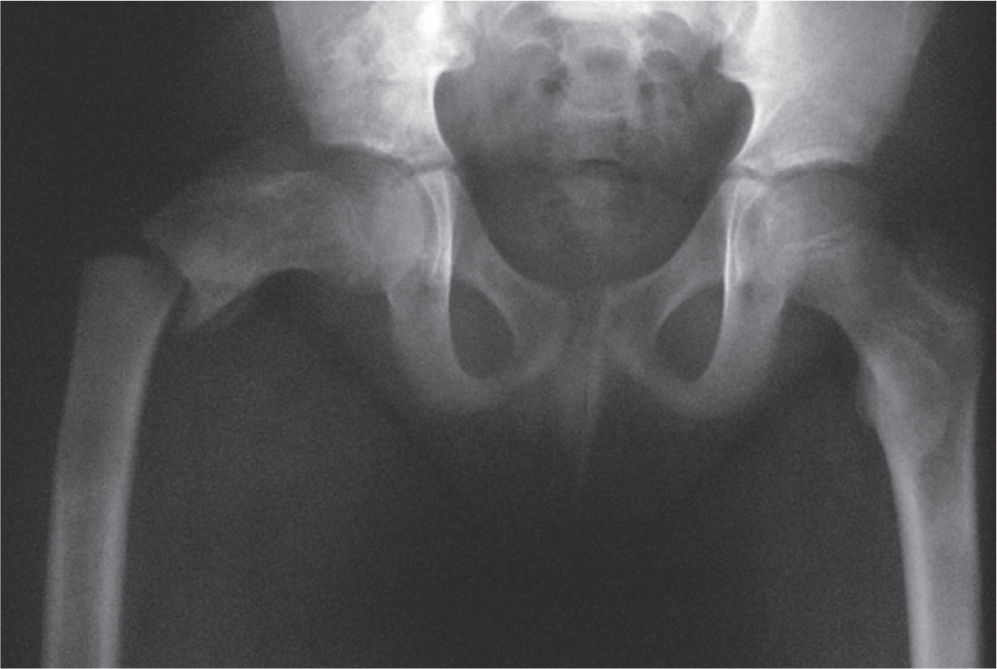
En el informe de la gammagrafía ósea corporal total tomada 2 meses antes del evento traumático, se observaron zonas hipointensas difusas en metáfisis proximales de los fémures, especialmente del derecho.
Fue en el marco de una complicación esperada de la enfermedad —como la ocurrencia de fracturas— que se pudo obtener, durante el procedimiento quirúrgico de fijación interna, datos clínicos intraoperatorios y la posterior caracterización en el ámbito histopatológico.
No obstante, el diagnóstico, según lo expresado por el departamento de patología, debe acompañarse de signos de expresión y comportamiento clínico debido a la similitud histológica con otro tipo de displasias fibrosas.
Así, podemos inferir que, para llegar a esta conclusión, es muy importante una buena historia clínica, incluso profundizando en antecedentes familiares de fracturas patológicas, que en ocasiones no se tiene en cuenta.
En el momento, el paciente tiene pendiente valoración por endocrinología para establecer un plan de manejo, sobre todo en cuanto a la administración oral de esteroides que puedan disminuir la progresión de la enfermedad; no obstante, su administración debe ser prudente, teniendo en cuenta la osteopenia que puede suceder a largo plazo luego de la toma crónica de estos medicamentos.
En el último control clínico y radiológico se ha observado mantenimiento de la reducción y alineación de extremos fracturarios, así como signos radiológicos tempranos de consolidación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
