




Idiopathic inflammatory myopathy is a heterogeneous group of autoimmune disorders, that share some common manifestations, such as muscle weakness and elevation of serum creatine kinase. However, classification of idiopathic inflammatory myopathy into an existing clinical subtype is not always possible. The case is a 25-year-old male with an unrecognized form of idiopathic inflammatory myopathy, the onset of which resembled amyotrophic lateral sclerosis. The paper includes differential diagnosis with amyotrophic lateral sclerosis and congenital myopathies, and response to corticosteroid therapy.
Las miopatías inflamatorias idiopáticas son un grupo heterogéneo de enfermedades autoinmunes que tienen ciertas manifestaciones clínicas como la debilidad muscular y el aumento del nivel de creatinina quinasa del suero en la sangre. Sin embargo, no siempre es posible hacer la clasificación de miopatía inflamatoria idiopática en el subtipo clínico existente. El caso clínico presentado describe a un varón de 25 años con una forma indeterminada de miopatía inflamatoria idiopática cuyo comienzo es parecido a la esclerosis lateral amiotrófica. En el trabajo se presenta el diagnóstico diferencial de esclerosis lateral amiotrófica y de miopatías hereditarias, así como la respuesta al tratamiento con corticosteroides.
Idiopathic inflammatory myopathy (IIM) is a heterogeneous group of autoimmune disorders, that share some common manifestations, such as muscle weakness and elevation of serum creatine kinase (CK).1 The presence of additional muscular, extramuscular and laboratory signs allows distinguishing a clinical phenotype of IIM.1 In some cases classification of a patient into a distinct phenotype is challenging.2
Case presentationA 25-years-old male was admitted to a hospital with complaints of weakness in the right hand and both feet, fasciculations in the muscles of the right shoulder, difficulties in the extension of the fingers of the right hand. The first symptoms appeared two years before the admission from disturbance of writing with the right hand, pain and limitation of movements in the right shoulder joint, weakness of muscles in the left hip. Since the onset of the disease a disruption of extension of the left index has been slowly progressing. The patient has lost 3kg since the onset of the disease. He had a history of frequent traumatizing of the right hand without seeing a doctor and trauma of the cervical spine, when a 30kg object had fallen on his neck. The patient had a history of professional sports (pole vaulting, boxing) in the past.
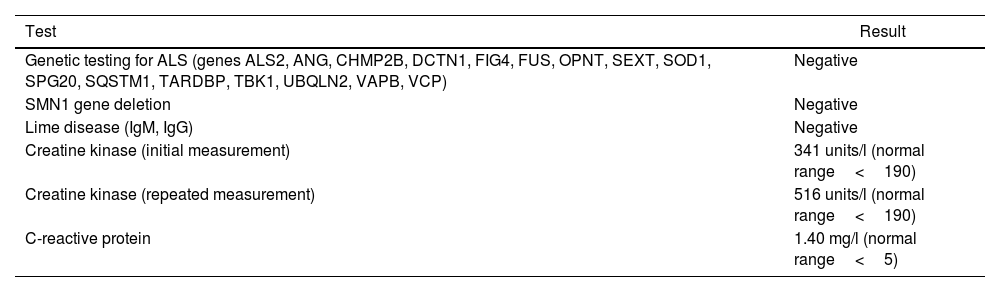
Shortly after the development of the first symptoms the patient was examined by neurologist. Neuromyography revealed neural injury (decreased conduction speed in the right radial nerve). Cranial and spine MRI revealed periventricular gliosis and posttraumatic deformation of processus spinosus Th1. ALS was suspected due to the suspected involvement of motoneurons of upper and lower limbs, presence of fasciculations3 and MRI findings. Genetic testing for ALS (Centre de Génétique Moléculaire et Chromosomique, Paris, France) did not reveal any known mutations that are associated with ALS (Table 1).4 Antibodies to Lime disease were negative. Serum CK was above the normal range. The patient was referred to a rheumatologist with suspicion of polymyositis.
Laboratory tests after initial manifestations.
| Test | Result |
|---|---|
| Genetic testing for ALS (genes ALS2, ANG, CHMP2B, DCTN1, FIG4, FUS, OPNT, SEXT, SOD1, SPG20, SQSTM1, TARDBP, TBK1, UBQLN2, VAPB, VCP) | Negative |
| SMN1 gene deletion | Negative |
| Lime disease (IgM, IgG) | Negative |
| Creatine kinase (initial measurement) | 341 units/l (normal range<190) |
| Creatine kinase (repeated measurement) | 516 units/l (normal range<190) |
| C-reactive protein | 1.40 mg/l (normal range<5) |
Patient's condition on the examination by rheumatologist: the patient had a normal body weight (height 193cm, weight 77kg, body mass index 20.7kg/m2), mild Raynaud's phenomenon in the hands, pronounced hypotrophy of the muscles of shoulder girdle, shoulders, forearms and hands (more on the right). A rash was absent and the patient denied the presence of a rash in the past. Swallowing of food and liquids was not disrupted, vocalization was not impaired. Palpation of the muscles of shoulder girdle, shoulder, forearm and hip was painful. The strength of shoulder girdle muscles was decreased. The patient was not able to stand up without help of hands from a chair. The fingers of the hands were in a slightly flexed position, Houston tabletop test was negative. Blood tests for antinuclear and myositis-specific antibodies, hepatitis B, C, human immunodeficiency virus were negative; serum CK was elevated, C-reactive protein was normal (Table 2). Muscle trephine biopsy (after magnetic resonance imaging scanning, from right quadriceps femoris muscle) showed muscle dystrophy and oedema. During two years of follow-up no evidence of neoplasm of any localization has been found.
Laboratory tests after hospitalization to rheumatology department.
| Test | Result |
|---|---|
| ANA | Negative |
| Autoanibodies: Ku, RNP, U1-nRNP, U3-nRNP, PM-SCL, Jo-1, Mi-1, Mi-2, MSA1, MSA2, HSAG5 | Negative |
| C-reactive protein | 1.55 mg/l (normal range<5) |
| Creatine kinase | 1003 units/l (normal range<190) |
| Anti-HBsAg (IgM, IgG) | Negative |
| Anti-HCV (IgM, IgG) | Negative |
| Anti-HIV (IgM, IgG) | Negative |
Note: ANA, antinuclear antibodies; HbsAg, hepatitis B surface antigen; HCV, hepatitic C virus; HIV, human immunodeficiency virus.
As the patient's condition continued to deteriorate he agreed to undergo a trial treatment with steroids (methylprednisolone 16mg/day). Three days after the onset of the treatment with steroids the patient reported diminution of pain and an increase of muscle strength in the hands and the legs (the patient began to stand up from a chair without help of the hands). Weakness of the right hand extensors and the fasciculations did not disappear. As the patient responded to steroids he was diagnosed with idiopathic inflammatory myopathy without further specification of the subset. The patient agreed to start a treatment with methotrexate (10mg/week) with close monitoring of his condition.
DiscussionThe classification of IIM has evolved in the last decades and now it includes five domains: dermatomyositis, overlap myositis, immune-mediated necrotizing myopathy, inclusion body myositis, and polymyositis.5 However, in some cases IIM have unique clinical and laboratory signs that do not allow to classify them into existing subset.2
The patient's condition did not fulfill the diagnostic criteria of ALS as he had other symptoms that could explain the patient's complaints (significant elevation of CK)3 and as the genetic testing was negative (Table 1). However, the diagnosis of ALS could not be ruled out definitely: a mild rise of CK may be observed in motor neuron diseases6 and a negative genetic testing does not exclude ALS, as the patient could be carrying mutations that are unknown yet. The majority of genetic alterations that could lead to muscular atrophy develop either early in childhood or after 40–50 years.6–8 Development of the symptoms in the patient early in the third decade of life suggested in favor of low probability of genetic background of the disease. However, testing for SMN1 deletion was performed in order to exclude type 4 spinal muscular atrophy, which may occur in early adulthood.9 Simultaneously, a good response to steroid treatment testified not in favor of ALS or congenital myopathy. There is no data that steroids are effective in patients with ALS, and their effect in patients with congenital myopathies is contradictory.10,11
The diagnosis of polymyositis was also doubtful, as the age of the disease onset was much younger than it is typical for polymyositis, skin manifestations were absent, muscle biopsy did not confirm immune inflammation in the muscles, there were nerve conduction abnormalities on neuromyography, and the test for autoantibodies was negative.12 Several symptoms allowed to suspect inclusion body myositis (IBM): asymmetrical muscle involvement, neuropathy on neuromyography, a mild increase of serum CK and absence of antinuclear or myositis-specific antibodies.1,13 But the pattern of muscular involvement in IBM (in particular, IBM with a higher probability affects finger flexors, than extensors)13 is different from what was observed in the patient. The patient responded to the treatment with steroids which is highly unlikely for IBM.1 The muscle biopsy did not reveal IBM-specific red-rimmed vacuoles.1 Therefore, the diagnosis of IBM also could not be confirmed.13
IIM is a rare group of diseases that has heterogeneous clinical manifestations. Moreover, a clinical profile of IIM changes over time14 and new forms are being discovered.2 Discovering new myositis-specific antibodies is in no doubt helpful in the diagnosis of IIM,15 but autoantibodies do not always accompany IIM.1 Furthermore, according to recent studies myositis specific antibodies may have low sensitivity and low positive predictive value, thus having a limited application in clinical practice for diagnostic purposes.5,16 All of the above makes challenging the definite diagnosis of IIM.
Strict adherence to guidelines may be not beneficial for a patient, as the delay in diagnosis will result in the delay in treatment. “Laboratory tests should be used to confirm your clinical diagnosis not make it”.1 Despite the presence in the patient of muscle weakness and CK elevation (the hallmark of IIM), additional manifestations were absent, making the diagnosis according to the criteria challenging.1 Given that the symptoms continued to worsen and CK levels increased it was decided to prescribe the trial treatment, which was successful. We suggest that patients with unknown forms of IIM may be prescribed with immunosuppressive treatment with a purpose to relieve symptoms, after a careful rule out of other conditions that may have similar manifestations.
ConclusionWe suggest that the patient in the presented case report suffers from an unknown subset of IIM, that had an atypical amyotrophic lateral sclerosis-like onset and bears clinical sings of both polymyositis and inclusion body myositis. Further monitoring of the patient's condition will allow making a conclusion about the effectiveness of immunosuppressive therapy.
Ethical disclosuresPresented study complies with the Declaration of Helsinki and was approved by Ethics Committee of Mechnikov Dnipropetrovsk Regional Clinical Hospital. Informed consent for analysis and publishing medical information was obtained from the patient.
Conflict of interestsAuthors declare no conflict of interests.
