



Inclusion body myositis is part of the group of inflammatory myopathies, representing 30% of this group of diseases, and is considered an orphan disease because its estimated prevalence is less than 5 per 10,000 inhabitants. It produces weakness and atrophy of the proximal and distal muscles. The pathophysiological mechanisms are mainly autoimmune, inflammatory, and degenerative. The cases are presented of two female patients who came to the emergency department due to progressive loss of upper and lower limb strength, and progressive asymmetric muscle weakness.
La miositis por cuerpos de inclusión hace parte del grupo de las miopatías inflamatorias, representando el 30% de este grupo de enfermedades, es considerada una enfermedad huérfana ya que se estima que su prevalencia es menor a 5 por cada 10.000 habitantes. Produce debilidad y atrofia de los músculos proximales y distales. Los mecanismos fisiopatológicos son principalmente autoinmunes, inflamatorios y degenerativos. Se presentan 2 casos de mujeres quienes acudieron a urgencias por pérdida progresiva de la fuerza en miembros superiores e inferiores, debilidad muscular asimétrica de curso progresivo.
The concept of inclusion body myopathy (IBM) was described for the first time in 1971 and adopted after the description of cases of patients with polymyositis resistant to conventional treatment (corticosteroids). The first diagnostic criteria for this disease were made known in 1995.1
IBM is a rare sporadic disorder, included within the group of idiopathic inflammatory myopathies. It is estimated that its prevalence is 5–9 cases per million adults, and that it is the most frequent acquired myopathy in people over 50 years of age (average age of onset, 60 years) with greater involvement in women.2
Regarding its pathophysiology, it is believed that aging associated with the accumulation and sedimentation of proteins in the intracellular environment plays a role in the pathogenesis, even though it appears that oxidative stress and the reaction to stress in the endoplasmic reticulum also contribute. The foregoing factors accumulatively lead to progressive degeneration and necrosis of the muscle. 3 main mechanisms of injury have been described: autoimmune, inflammatory and degenerative. Cytotoxic T cells play an important role, since they invade and destroy the muscle fibers, mainly those that are vacuolated or with amyloid deposits, and additionally generate neurodegenerative damage.2–5
The main clinical manifestation is generalized weakness of insidious course, of both distal and proximal involvement, with predominance of the latter, expressed by the patient as an inability to get up from the chair or as frequent falls. On physical examination, the main characteristic is weakness of the flexor muscles of the fingers distally.6,7
Clinical findings that differ from other diseases of the same group (dermatomyositis or polymyositis) are distal and asymmetric commitment, presence of myalgia of mild intensity, greater than expected atrophy according to the time of the clinical picture and, rarely, severe dysphagia.8
The finding of elevated muscle enzymes is the main laboratory abnormality in these patients. A slight elevation of creatinine kinase less than 10 times the normal value is the main differentiator from other inflammatory myopathies. Electromyographic findings correspond to inflammatory myopathy. Antibodies specific for myositis are often absent in IBM. The typical pathological characteristics of this entity are: rimmed vacuoles with atrophic or normal muscle fibers, inflammatory infiltrates and CD8+ T lymphocytes.9
The first accepted clinical criteria were those proposed by Griggs in 1995; later, the European Neuromuscular Centre in 2011 defined some criteria to classify the disease into 2 categories: clinical-pathological IBM and clinical IBM.1
The optimal treatment of IBM is currently unknown and most of the interventions have had limited benefits, for which the use of immunosuppressive therapy is compulsory, with an unfavorable prognosis when compared with other inflammatory myopathies.10
We present 2 cases of women over 50 years of age, who arrived to the emergency department due to loss of muscle strength in the upper and lower limbs, asymmetric, of progressive course, in which a histopathological and clinical diagnosis of IBM was achieved. Given that this entity is rare in our environment, it was decided to report both cases in the literature.
Clinical case 1A 55-year-old woman, housewife, right-handed, with a history of systemic arterial hypertension and controlled primary hypothyroidism, who arrived to the emergency department for a clinical picture of 4 months of evolution consisting in progressive loss of muscle strength in the pelvic and shoulder girdle. The weakness progressed insidiously in the past 20 days, until she required assistance with her basic activities of daily life. In the review by systems, it was documented in the last month an unintentional weight loss of about 10 kg, associated with progressive hyporexia. The patient denied diaphoresis, febrile peaks, cutaneous lesions and other concomitant symptoms. There was no positive family history for autoimmune diseases. On the physical examination on admission, she was unable to raise her arms above her shoulders and to get up from a chair; the muscle strength was 3/5 in all the extremities, except in the left lower limb, in which it was 2/5. The osteotendinous reflexes were normal, and there were no sensory alterations.
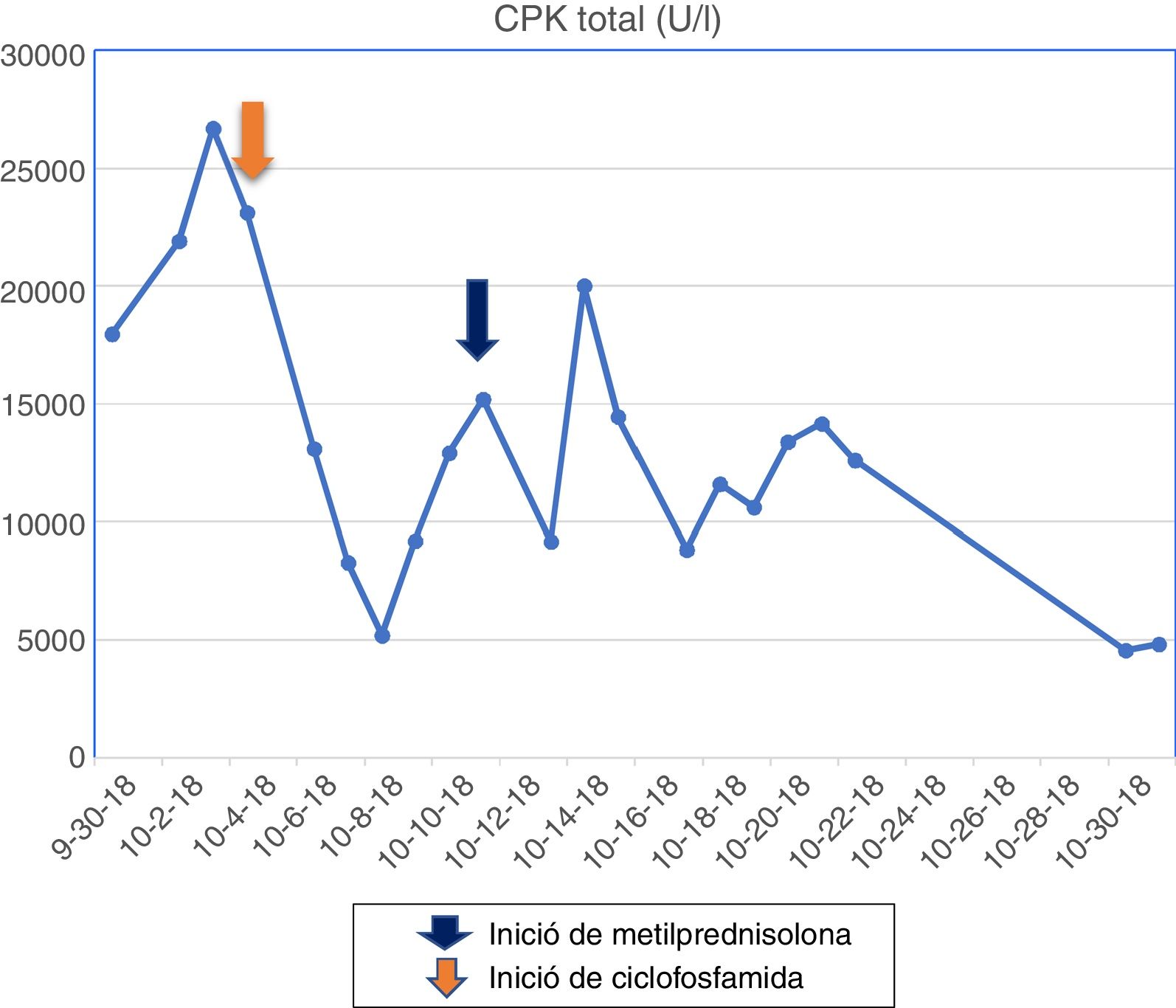
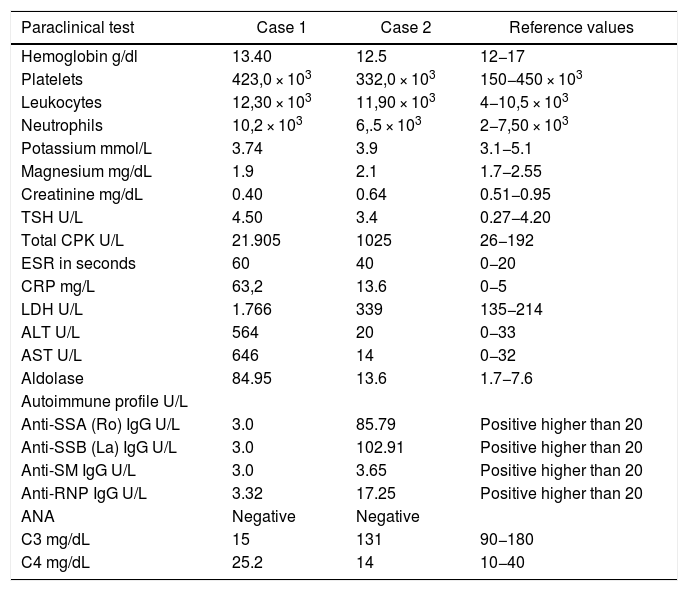
The patient had a chronic formulation of 20 mg of lovastatin for 2 years as primary prevention, without knowing the reasons for its prescription, which in the context of the myopathy was withdrawn. The paraclinical tests on admission are shown in Table 1 and the changes in the total CPK curve are seen in Fig. 1.
Serological characteristics of the treated patients.
| Paraclinical test | Case 1 | Case 2 | Reference values |
|---|---|---|---|
| Hemoglobin g/dl | 13.40 | 12.5 | 12−17 |
| Platelets | 423,0 × 103 | 332,0 × 103 | 150−450 × 103 |
| Leukocytes | 12,30 × 103 | 11,90 × 103 | 4−10,5 × 103 |
| Neutrophils | 10,2 × 103 | 6,.5 × 103 | 2−7,50 × 103 |
| Potassium mmol/L | 3.74 | 3.9 | 3.1−5.1 |
| Magnesium mg/dL | 1.9 | 2.1 | 1.7−2.55 |
| Creatinine mg/dL | 0.40 | 0.64 | 0.51−0.95 |
| TSH U/L | 4.50 | 3.4 | 0.27−4.20 |
| Total CPK U/L | 21.905 | 1025 | 26−192 |
| ESR in seconds | 60 | 40 | 0−20 |
| CRP mg/L | 63,2 | 13.6 | 0−5 |
| LDH U/L | 1.766 | 339 | 135−214 |
| ALT U/L | 564 | 20 | 0−33 |
| AST U/L | 646 | 14 | 0−32 |
| Aldolase | 84.95 | 13.6 | 1.7−7.6 |
| Autoimmune profile U/L | |||
| Anti-SSA (Ro) IgG U/L | 3.0 | 85.79 | Positive higher than 20 |
| Anti-SSB (La) IgG U/L | 3.0 | 102.91 | Positive higher than 20 |
| Anti-SM IgG U/L | 3.0 | 3.65 | Positive higher than 20 |
| Anti-RNP IgG U/L | 3.32 | 17.25 | Positive higher than 20 |
| ANA | Negative | Negative | |
| C3 mg/dL | 15 | 131 | 90−180 |
| C4 mg/dL | 25.2 | 14 | 10−40 |
ANA: antinuclear antibodies; anti-RNP: anti-ribonucleoprotein antibodies; anti-SM: anti-Smith antibodies; anti-SSA (Ro): anti-Ro antibodies; C3: serum complement C3; C4: serum complement C4; g/dl: grams/deciliter; mg/dl: milligrams/deciliter; mg/l: milligrams/liter; mmol/l: millimoles/liter; U/l: unit.

The muscle MRI of the left lower limb evidenced fat infiltration in the muscle bundles, in addition to muscle atrophy, especially in the femoral biceps, semimembranosus, semitendinosus, gracilis and sartorius muscles, vastus medialis, vastus lateralis, vastus intermedius and part of the quadriceps. The electromyography reported a pattern of myopathic involvement. Pulses of methylprednisolone for 5 days were started, and later pulses with cyclophosphamide. Despite the decline in the levels of muscle enzymes, there was no clinical response.
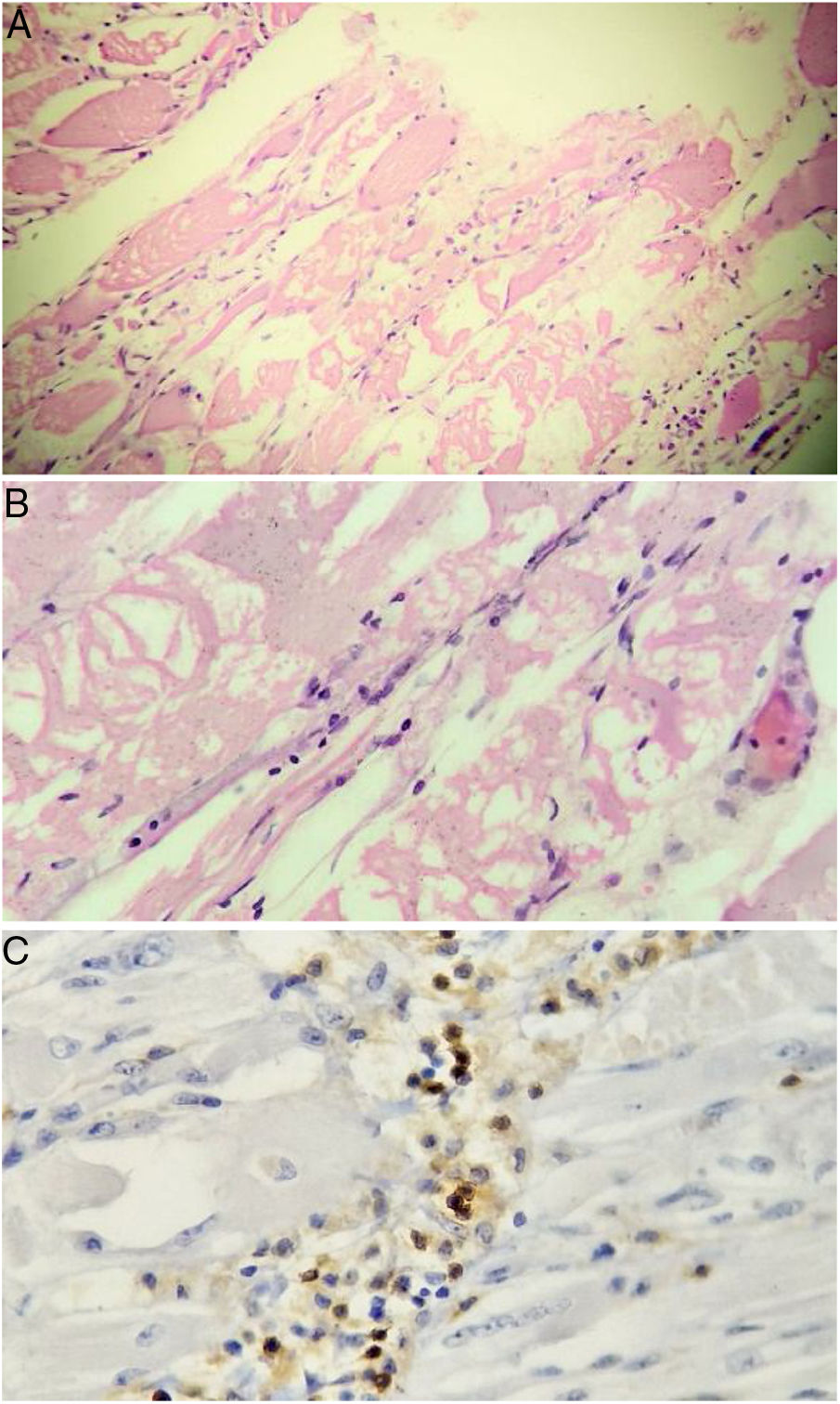
Taking into account the loss of 10 kg and considering that inflammatory myopathies can be associated with cancer, extension studies were requested in order to rule out a neoplastic disease. Abdominal and pelvic axial tomography, endoscopy of the upper gastrointestinal tract, diagnostic breast ultrasound, colonoscopy, and thyroid ultrasound were performed, which were negative for masses, lymphadenopathies and metastases. Tumor markers were also obtained: ACE-CEA (carcinoembryonic antigen), CA 125 (ovarian cancer antigen), CA 19-9 (gastrointestinal cancer antigen), which were also negative. Based on the magnetic resonance imaging report, it was decided to carry out a muscle biopsy: histologically, changes suggestive of myofibrillar degeneration and phagocytosis with significant lymphocytic infiltration were evidenced (Fig. 2). With the foregoing, the presence of inclusion body myositis was confirmed.
 are observed. Lymphocytic inflammatory infiltrate, both endomysial and perivascular. Immunoperoxidase markers positive for CD20 in the inflammatory infiltrates.")
Muscle biopsy. Rounded atrophic muscle fibers with a myopathic appearance of perifascicular predominance, accompanied by myopathic changes (myofibrillar degeneration and phagocytosis with regeneration) are observed. Lymphocytic inflammatory infiltrate, both endomysial and perivascular. Immunoperoxidase markers positive for CD20 in the inflammatory infiltrates.
Multidisciplinary management was indicated. In the course of her evolution the patient presented increased weakness and dysphagia, initially for solid foods and later for liquids; she also presented sialorrhea and cough with the ingestion of food. A cinevideo swallowing exam was requested, which reported a severe swallowing alteration in the oral and pharyngeal phases, constant and repetitive reflux towards the nasopharynx, for which a percutaneous gastrostomy was proposed, which the patient did not accept. After 30 days of hospitalization in which there was deterioration in the clinical picture of the patient, characterized by generalized weakness in the neck and the 4 extremities, with muscle strength 1/5, and inability to mobilize against gravity, there was no successful response to the physical rehabilitation therapy. The patient, in the company of her family nucleus, signed a voluntary departure from the institution, despite the clinical evolution and her condition at discharge.
Clinical case 2A 61-year-old female patient, right-handed, with a history of mumps, Sjögren’s syndrome (SS) and Raynaud’s phenomenon, who consulted for a clinical picture of several months of evolution consisting of progressive generalized proximal weakness, predominantly in the lower limbs with slow evolution until gait impairment, associated with progressive weight loss of approximately 6 kg. In the directed interrogation she reported sicca symptoms and Raynaud’s phenomenon. The patient denied skin changes, hands swelling, and dysphagia, among others. Without relevant family antecedents. On physical examination, the decrease in strength in the 4 extremities, of proximal predominance of 4/5, without alteration in sensitivity or osteotendinous reflexes drew attention. In addition, it was found distal pallor in the hands, consistent with Raynaud’s phenomenon.
The initial paraclinical tests are shown in Table 1. Other additional paraclinical tests were: rheumatoid factor 159.6 IU/mL (high), serum protein electrophoresis with polyclonal γ peak, echocardiogram and chest X-ray within normal parameters.
Due to complaint, in addition to referred low back pain, it was performed a magnetic resonance imaging of the lumbosacral spine, in which signs of depression fractures of the inferior articular surfaces of L3 and L4 were evidenced with bone marrow edema; in addition to multiple lumbar discopathy and disk protrusion which contacted the dural sac and the S1 nerve roots. The foregoing was suggestive of osteoporosis and lumbar discopathy.
Electromyography and nerve conduction study of 3 limbs was performed, which reported a pattern of inflammatory muscle disease. The muscle biopsy documented atrophic perifascicular muscle fibers, with myofibrillar degeneration and phagocytosis, with presence of vacuoles and perivascular lymphocytic infiltrate compatible with IBM. Management with pulses of methylprednisolone and cyclophosphamide, in addition to physical therapy was indicated. Currently, she is receiving the fourth dose of cyclophosphamide, with a favorable evolution, partial improvement of strength in the 4 limbs and a gradual decrease in muscle enzymes.
DiscussionInflammatory myopathies are a group of systemic diseases that produce a clinical picture of muscle weakness, elevation of muscle enzymes, myopathic findings on electromyography and inflammatory pattern in muscle biopsy.2,3 They are classified according to their presentation, onset, extramuscular manifestations, histological characteristics, and response to treatment. The main types are: dermatomyositis, inclusion body myositis, polymyositis, and necrotizing autoimmune myopathy.1,6,7,10
We describe 2 cases of patients with no family history, with insidious clinical pictures of weakness that fulfilled 3 Bohan and Peter criteria, which increases the clinical possibility for inflammatory myopathy; however, due to high suspicion of IBM, Griggs criteria1 were applied, by performing muscle biopsies which were consistent with an inclusion body myositis.11–13 In both patients, the poor response to the established medical treatment and the initial asymmetrical presentation, and in the first case, the rapid evolution to distal commitment were striking. All these characteristics have been described as predictors of poor prognosis of the IBM and, in addition, they are contrary to the course of polymyositis or dermatomyositis in which, in general, a favorable clinical course can be seen after the start of treatment and in which the commitment is usually of proximal predominance. For this reason, the IBM becomes a therapeutic challenge, since it requires the onset of an immunomodulatory therapy without optimal effectiveness.10
In a series of 30 cases of IBM published in Brazil, with a 30-year follow-up,14 it was found that the main manifestations, besides the involvement of the muscle strength, were dysphagia, weight loss and cardiac alterations, being arthralgia, respiratory symptoms and dysphonia less common. In both reported cases, the patients manifested weight loss, however, only the first reported dysphagia. None of the patients exhibited cardiac symptoms.
Dysphagia has been related as a complication of IBM, in which it has been found that about 38% of patients can manifest it.15 Another important complication is respiratory failure. Despite an exhaustive search, no clinical or paraclinical manifestation of neoplastic etiology was found. Although IBM is a rare cause of paraneoplastic syndrome, as exposed by Dardis et al., it is important the search for malignancy, mainly with thyroid ultrasound and tomographic scanning, due to post mortem findings of malignant disease in these patients.16
The correlation of IBM with SS has been found reported in the literature. A case report published by Misterska-Skora et al.17 shows the case of a 57-year-old female patient with dry symptoms, Raynaud's phenomenon, and proximal and distal muscle weakness of the 4 limbs, who was finally diagnosed with IBM and SS, with clinical improvement after the start of the treatment, and in whom the concomitant therapy for the SS, as it was concluded, could have improved the prognosis of the muscle disease, a clinical picture similar to that reported in our second case.
The diagnosis of inclusion body myositis can be difficult, since the clinical presentation has a slow evolution and in the first years it can be nonspecific. Given the foregoing, a high index of suspicion is required, as well as a comprehensive association of the clinical history, the levels of muscle enzymes and complementary studies such as biopsy and electromyography.11,12 It is necessary to complement the study of these patients to rule out the coexistence of autoimmune diseases and malignancy. Cytoplasmic antibodies against Mi-2 antigens, anti-tRNA synthetase, anti-SRP, the transcription factor 1-γ, and the melanoma differentiation-associated gene 5 are useful for this purpose. In none of the 2 cases presented was possible to take samples for these antibodies.1,3,5,9
Among the differential diagnoses of the inflammatory myopathies, we must consider the metabolic myopathies: glycogen storage disorders (McArdle disease, Pompe disease), genetic myopathies: shoulder girdle muscular dystrophy, facioscapulohumeral dystrophy, myotonic dystrophy, myofibrillar myopathies, congenital myopathies, and skeletal muscle channelopathies. Neurological diseases: amyotrophic lateral sclerosis, spinobulbar muscular atrophy (Kennedy’s disease), peripheral nerve hyperexcitability and radiculopathies.18
Other acquired diseases with myopathic manifestations are: infectious and post-infectious myositis, endocrinopathies (hyper- or hypothyroidism, acromegaly, Cushing’s syndrome, Addison’s disease, vitamin D deficiency, hyper- or hypocalcemia, hypokalemia), pyonecrosis and trauma. The use of drugs or toxins involved in myotoxicity and associated with the use of statins, fibrates, colchicine, hydroxychloroquine, zidovudine, cocaine, alcohol, penicillamine, among others, should also be considered.18
The muscle biopsy plays an important role as part of the diagnostic process in the evaluation of a patient with a neuromuscular condition and is essential for the confirmation of the IBM, in which signs of chronicity such as hypertrophic, atrophic fibers, partitions, central nuclei and fat infiltration are usually described. The «major» histological findings are multifocal lymphocytic infiltrate that invades non-necrotic fibers, vacuoles in cells not invaded by lymphocytes (these vacuoles of rimmed type contain basophilic granular deposits) and Congo-red positive amyloid deposit. The finding of ragged-red fibers and negative cytochrome-oxidase is frequent, as a consequence of mitochondrial dysfunction.5,9,19 In both reported biopsies of the patients, multifocal lymphocytic infiltrate and myofibrillar phagocytosis with the presence of vacuoles were consistent, compatible with that is described in the literature for this entity.
The prognosis of the disease is ominous, taking into account the progressive and disabling course. It is emphasized that these patients require interdisciplinary management which includes the participation of neurology, internal medicine, psychiatry, psychology, physiatry, physical rehabilitation and respiratory therapy, among others.10
ConclusionInclusion body myositis is a diagnosis to be considered in patients with proximal and distal asymmetric weakness, with low titers of creatinine kinase and in those who do not respond to conventional therapy. It is important to reach an early diagnosis in order to avoid complications that could worsen their prognosis. Treatment should include immunosuppressive therapy, psychological support and optimal physical rehabilitation in order to impact the quality of life of the patients.
FundingNone.
Conflict of interestThere is no conflict of interest.
Please cite this article as: Bolaños-Toro F, Arias-Jaramillo D, Aun-Mejía M, Castro-Henao J, Fernández-Ávila DG, Saldarriaga-Rivera LM. Miopatía por cuerpos de inclusión: un diagnóstico diferencial que considerar en pacientes con miopatía refractaria a inmunosupresores. Reporte de 2 casos. Rev Colomb Reumatol. 2021;28:300–305.
