





El penfigoide ocular cicatrizal (POC) es una enfermedad crónica, inmunomediada ampollar, mucosinequiante, comprendida dentro del espectro de penfigoides membranosos mucocutáneos (PMM). El diagnóstico es, con frecuencia, oftalmológico, pero debido al carácter autoinmune de la patología, requiere el abordaje en conjunto con reumatólogos e inmunólogos. El objetivo de esta revisión narrativa fue explorar la evidencia disponible en la literatura, desde el año 2000 hasta el 2020, en lo que respecta a sus manifestaciones clínicas, diagnóstico y tratamiento. La presentación clínica varía ampliamente, desde casos leves con progresión lenta de años de evolución hasta casos severos con evolución tórpida y rápidamente progresiva a la fibrosis, refractarios a múltiples tratamientos. Una evaluación completa del paciente ayudará a guiar el diagnóstico. El estándar de oro diagnóstico es la biopsia conjuntival con inmunofluorescencia directa, si bien en ocasiones puede diagnosticarse por la clínica característica. El tratamiento es local y sistémico de acuerdo con su severidad y evolución. En los últimos 20 años, la evidencia sobre los tratamientos tópicos y sistémicos corresponde en su mayoría a estudios observacionales y experimentales no controlados. Los métodos de tratamiento inmunomoduladores han permitido preservar la visión y, en muchos casos, prevenir secuelas. La evolución está ligada al diagnóstico temprano y a los tratamientos disponibles, por lo que es fundamental el conocimiento de esta patología, los métodos diagnósticos y los tratamientos inmunomoduladores e inmunosupresores.
Ocular cicatricial pemphigoid (POC) is a chronic, immune-mediated, bullous, cicatricial disease within the spectrum of mucocutaneous membranous pemphigoids (PMM). Although the diagnosis is often ophthalmological, due to the autoimmune nature of the pathology, it requires a joint approach with rheumatologists and immunologists. The objective of this narrative review was to explore the evidence available in the literature from 2000 to 2020 with respect to clinical manifestations, diagnosis, and treatment. The clinical presentation varies widely, from mild cases with slow progression of years of progression, to severe cases with a torpid and rapidly progressive evolution to fibrosis, refractory to multiple treatments. A complete evaluation of the patient will help guide the diagnosis. The gold standard for diagnosis is conjunctival biopsy with direct immunofluorescence, although on occasions it can be reached if the symptoms are characteristic. Treatment is local and systemic according to its severity and evolution. The evidence on topical and systemic therapeutics is obtained mainly from uncontrolled observational and experimental studies. Immunomodulatory therapy has made it possible to preserve vision and, in many cases, prevent sequelae. The evolution is linked to the early diagnosis and immunosuppressive treatment, so it is essential to be aware of this disease, the diagnostic methods, as well as the immunomodulating and immunosuppressive therapies available.
El penfigoide ocular cicatrizal (POC) es una enfermedad crónica, inmunomediada ampollar, comprendida dentro del espectro de penfigoides membranosos mucocutáneos (PMM). En el PMM cualquier membrana mucosa puede estar afectada, como la tráquea, la laringe, el esófago, la vagina, el ano o la uretra, mientras que cuando el compromiso es fundamentalmente de la conjuntiva ocular recibe el nombre de POC, el cual se evidencia en un 32-48% de los PMM1–5.
Afecta a aproximadamente 1,3 a 2 personas por millón de habitantes, con una incidencia mayor entre los 50-60 años, si bien se han reportado variaciones de edad entre los 20 y los 80 años y una predominancia femenina 2-3:1. Distintas series estiman una incidencia anual de 1 entre 15.000 y 46.000 pacientes oftalmológicos. La prevalencia estimada en Estados Unidos fue de 1/20.000 a 1/60.000 en consultas oftalmológicas, y en población hispana el número de pacientes reportados es escaso, por lo que se sospecha una baja prevalencia o un subdiagnóstico o subreporte de la patología3–13.
Si bien la etiopatogenia del POC no se conoce con exactitud, se plantea que podrían estar involucrados factores genéticos y ambientales, produciéndose una respuesta de hipersensibilidad tipo II. Se caracteriza por autoanticuerpos dirigidos a los sitios de anclaje entre las células epiteliales y la membrana basal, siendo el sitio principal de afectación la membrana basal epitelial, a diferencia de otras enfermedades ampollosas como los pénfigos. La relación antígeno-anticuerpo da como resultado la iniciación de la cascada inflamatoria, de modo que se produce una inflamación aguda y crónica con formación de cicatrices. Existe un depósito en la membrana basal epitelial de IgA, IgG, IgM y c3 (IgG e IgA con mayor frecuencia) y fragmentos del complemento. Este depósito es lineal, a diferencia de otras entidades que tienen un depósito granular. Se han descripto como antígenos involucrados en las proteínas hemidesmosómicas, lamininas, integrinas y colágenas3,4,6–8.
En la fase aguda se observan eosinófilos y neutrófilos, mientras que en la fase crónica se detectan predominantemente linfocitos y fibroblastos a nivel subepitelial. Diversas citoquinas se han observado en cantidades abundantes en la conjuntiva, tales como IL-1, IL-2, IL-4, IL-5, IL-6, IL-13, IL-17, TNFα, TNFγ y factores de crecimiento de colonias. La IL-13, el TNFα y el factor de crecimiento β1 presentan un efecto profibrótico y proinflamatorio, activando y perpetuando la sobrevida de fibroblastos con el consecuente depósito de colágeno. En muestras de lágrimas se han detectado niveles elevados de IL-8, metaloproteinasas y mieloperoxidasas9–11.
Con frecuencia el diagnóstico es oftalmológico, pero debido al carácter autoinmune de la patología requiere el abordaje en conjunto con reumatólogos e inmunólogos. La comprensión del POC ha cambiado en forma significativa en las últimas décadas. El aporte de las ciencias básicas ha sido clave en la descripción de los procesos que participan en la respuesta inmune. La terapéutica inmunomoduladora ha permitido preservar la visión y, en muchos casos, prevenir las consecuencias fatales para esta en el curso de la enfermedad. La evolución está ligada al diagnóstico temprano y oportuno y al tratamiento inmunosupresor3,4,6–8, por lo que es sustancial el conocimiento de esta patología y de la evidencia disponible. La progresión de la enfermedad puede llevar a discapacidad funcional, como la ceguera corneal, con afectación de la calidad de vida. Por su parte, los costos socioeconómicos de pacientes con discapacidad, sumados a los tratamientos inmunosupresores, refuerzan la necesidad de un diagnóstico y un tratamiento precoces14–16.
Este artículo tiene como objetivo realizar una revisión de la evidencia publicada en los últimos 20 años, desde el 2000 al 2020, en relación con las manifestaciones clínicas, el diagnóstico y los tratamientos disponibles en pacientes con penfigoide ocular cicatrizal en diferentes estadios evolutivos, con el fin de actualizar y familiarizar a médicos en general sobre el tema.
Materiales y métodosRevisión narrativa de la literatura. Se realizó una búsqueda de literatura en PubMed, Cochrane Library, Lilacs, Epistemonikos y SciELO sobre la evidencia disponible en los últimos 20 años, desde el 2000 al 2020 en relación con las manifestaciones clínicas, el diagnóstico y los tratamientos disponibles en pacientes con penfigoide ocular cicatrizal, en diferentes estadios evolutivos. Se incluyeron artículos completos, con diseños de estudio experimentales, observacionales, revisiones, series de casos y reportes de casos. Se excluyeron resúmenes, artículos repetidos y en idiomas diferentes al español y al inglés. La estrategia de búsqueda consistió en la búsqueda de términos controlados en la Biblioteca Virtual de Salud, Descriptores de Salud (DeCS) o Medical Subject Readings, en inglés y en español, con operadores booleanos:
[Penfigoide Benigno de la Membrana Mucosa / Pemphigoid, Benign Mucous Membrane / Penfigoide Mucomembranoso Benigno / Ocular cicatricial pemphigoid / Penfigoide ocular cicatrizal / Síndromes de Ojo Seco / Dry Eye Syndromes]] AND [Terapéutica / Therapeutics OR Dapsona / Dapsone OR Sulfasalazina / Sulfasalazine OR Pentoxifilina / Pentoxifylline OR Hidroxicloroquina / Hydroxychloroquine OR Metotrexato / Methotrexate OR Azatioprina / Azathioprine OR Leflunamida / Leflunomide OR Tacrolimus / Tacrolimus Ácido Micofenólico / Mycophenolic Acid OR Ciclofosfamida / Cyclophosphamide OR Rituximab / Rituximab OR Inmunoglobulinas Intravenosas / Immunoglobulins, Intravenous OR Adalimumab / Adalimumab OR Etanercept / Etanercept OR Infliximab / Infliximab OR Certolizumab Pegol / Certolizumab Pegol OR golimumab/golimumab].
[Penfigoide Benigno de la Membrana Mucosa / Pemphigoid, Benign Mucous Membrane / Penfigoide Mucomembranoso Benigno / Ocular cicatricial pemphigoid / Penfigoide ocular cicatrizal] AND Diagnóstico / Diagnosis OR Técnicas de Diagnóstico Oftalmológico / Diagnostic Techniques, Ophthalmological.
[Penfigoide Benigno de la Membrana Mucosa / Pemphigoid, Benign Mucous Membrane / Penfigoide Mucomembranoso Benigno / Ocular cicatricial pemphigoid / Penfigoide ocular cicatrizal] AND Manifestaciones Oculares / Eye Manifestations.
Se amplió la búsqueda con términos no controlados en la literatura gris mediante Google Schoolar y OpenGray, tales como: tofacitinib, baricitinib, tocilizumab, ciclosporina a, tracolimus y guías. En estos casos se incluyeron dos resúmenes.
La evaluación de la evidencia se basó en Lancet 200217, donde se estipula, de acuerdo con el diseño de estudio, que la calidad de evidencia I es la proveniente de al menos un estudio clínico aleatorizado, controlado, bien diseñado; la II-1 es aquella obtenida de un estudio clínico controlado no aleatorizado; la II-2 es la obtenida de un estudio de casos y controles o de cohorte bien diseñado, preferentemente de más de un centro o grupo de estudio; la II-3 es la obtenida de múltiples series de casos con o sin intervención y de resultados importantes de estudios experimentales no controlados; y la III es la obtenida de opinión de expertos, basadas en experiencia clínica, estudios descriptivos o reportes de comités de expertos.
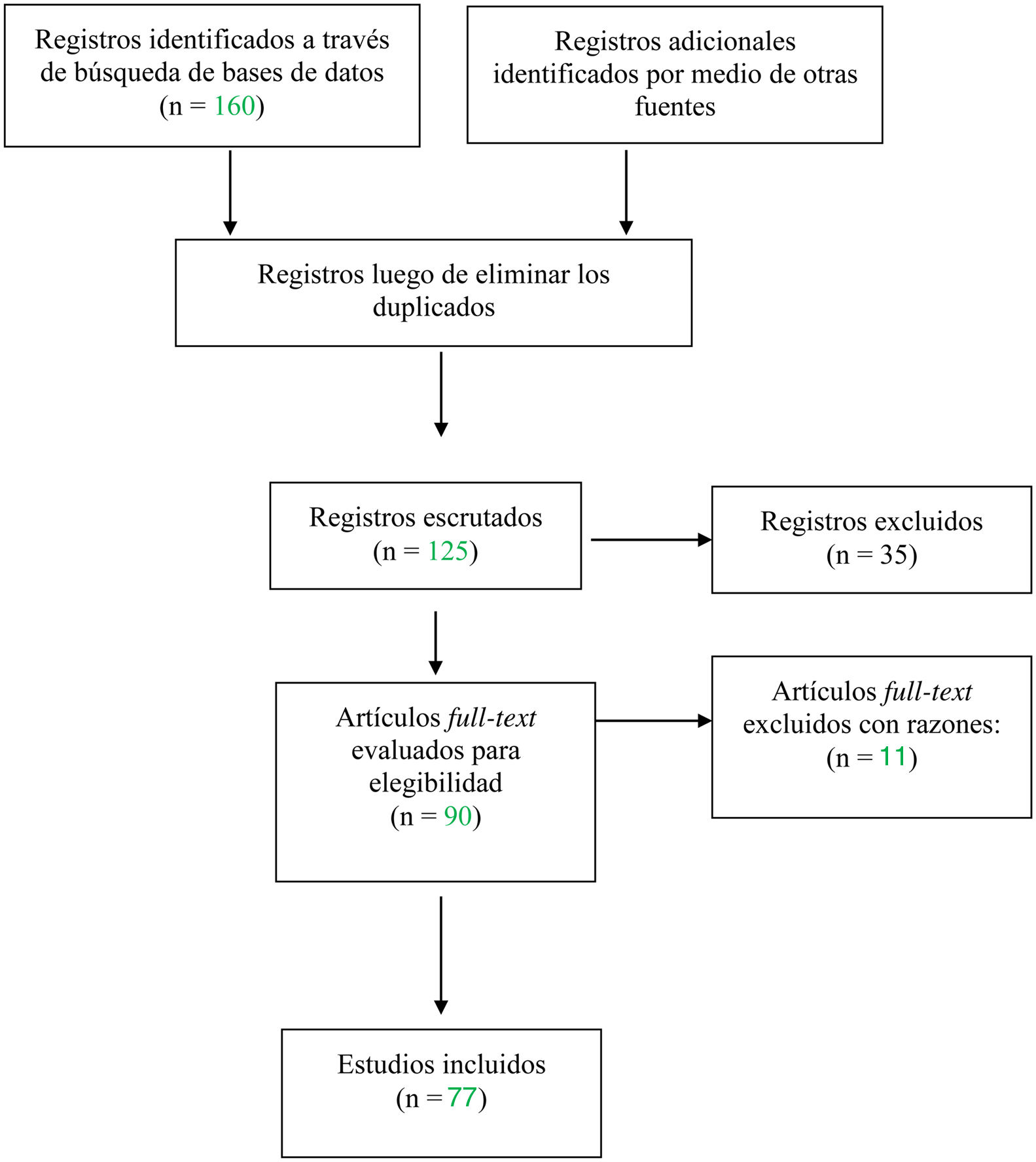
ResultadosFlujograma de búsqueda y selecciónManifestaciones clínicas
La presentación clínica varía ampliamente, desde casos leves con progresión lenta de años de evolución hasta casos severos con evolución tórpida y rápidamente progresiva a la fibrosis, refractarios a múltiples tratamientos. La mayoría de los pacientes presenta una evolución lenta, con exacerbaciones ocasionales3,4,6–8.
El comienzo puede ser unilateral o bilateral; generalmente, compromete ambos ojos. Cuando es unilateral, el ojo contralateral suele afectarse dentro de los 2 años. El POC suele manifestarse inicialmente como conjuntivitis recurrente, ojo rojo o hiperemia, ardor, dolor, prurito, lagrimeo, fotofobia y sensación de cuerpo extraño. Puede evolucionar a una queratopatía progresiva con neovascularización y conjuntivalización de la córnea, posteriormente aparece el simbléfaron, definido como una adherencia cicatrizal que se produce entre la conjuntiva del párpado y el globo ocular, adherencia que es posible visualizar deprimiendo el párpado inferior y observando pliegues verticales en la conjuntiva. En estadios más avanzados evoluciona a anquilobléfaron, que es la fusión de los bordes de los párpados, generalmente en la zona más próxima al canto externo y con desaparición del saco conjuntival. En el POC se observa ojo seco, de severidad variable de acuerdo con el estadio. Las tres capas de la película lagrimal se ven alteradas (mucinosa, acuosa y lipídica). En formas tardías disminuyen o desaparecen las células caliciformes del epitelio conjuntival, las cuales secretan mucina y contribuyen a mantener la capa lagrimal. Por otro lado, la cicatrización conduce a la disfunción de las glándulas de Meibomio, fibrosis de sus orificios y consecuente alteración de la capa lipídica, siendo esta la que más afecta la evaporación y estabilidad de la lágrima3,4,6–8,11–13,18.
La cicatrización conjuntival puede llevar a entropión (inversión del párpado), triquiasis y distriquiasis (eversión y crecimiento anormal de las pestañas, secundarios a la cicatrización del margen del párpado) y lagoftalmos (imposibilidad de cerrar completamente los párpados, con dificultad de distribución de la lágrima por la superficie ocular). La exposición ocular a su vez lleva al daño corneal, con erosiones que junto a la sequedad, la distriquiasis y los lagoftalmos fomentan la aparición de pannus, seudopterigión y una mayor cicatrización con alteración de la visión en grado variable e incluso ceguera. A su vez, la sobreinfección bacteriana también es frecuente. En casos muy severos puede ocurrir una perforación corneal. En la práctica oftalmológica, el cuadro que se presenta inicialmente con mayor frecuencia y que hace sospechar el diagnóstico es una conjuntivitis crónica, con sequedad ocular, pequeñas cicatrices conjuntivales, distriquiasis y estrechamiento u oclusión de los puntos lagrimales3,4,6–8,10–13,18–20.
Se ha sugerido que aun en casos en los que la conjuntiva se observa blanca y sin inflamación, pueden existir infiltrados inflamatorios subyacentes no observables (inflamación blanca, inflamación escondida), lo que podría continuar estimulando el proceso fibrótico11,20,21.
Existen diversas clasificaciones: la de Foster1 se basa en signos clínicos de progresión, mientras que la de Mondino y Brown2 se basa en la evaluación de la profundidad del fórnix inferior. El compromiso suele ser asimétrico, pueden observarse distintos estadios en ambos ojos (tabla 1).
Clasificación
| Clasificación de Foster | |
| Estadio 1 | Presencia de fibrosis subepitelial e inflamación conjuntival |
| Estadio 2 | Presencia de retracción de la conjuntiva con acortamiento de los fórnices |
| Estadio 3 | Presencia de simbléfaron |
| Estadio 4 | Presencia de anquilobléfaron |
| Clasificación de Mondino y Brown | |
| Estadio 1 | Pérdida de la profundidad del fórnix entre 0 y 25% |
| Estadio 2 | Pérdida de la profundidad del fórnix entre 25 y 50% |
| Estadio 3 | Pérdida de la profundidad del fórnix entre 50 y 75% |
| Estadio 4 | Pérdida de la profundidad del fórnix entre 75 y 100% |
También es importante la clasificación de acuerdo con la severidad: se considera leve la presencia de cicatrización y fibrosis subconjuntival; moderada, el acortamiento del fondo de saco inferior del ojo; grave, la presencia de simbléfaron con compromiso horizontal; y muy grave, la observación de anquilobléfaron. La actividad se evalúa por la presencia de fotofobia, dolor ocular, vascularización e hiperemia conjuntival, edema y cicatrización progresiva7.
Descripción oftalmológica detallada de los distintos estadios1,2Estadio 1: incluye inflamación conjuntival, secreciones, sectores epiteliales que tiñen con Rosa de Bengala y fibrosis subepitelial. Esta fibrosis es muy sutil y se observa únicamente con lámpara de hendidura, aparecen como estrías muy finas que rodean los vasos, y estas estrías de tejido conectivo anormal son las que finalmente se contraen y van a causar la retracción conjuntival.
Estadio 2: se caracteriza por la retracción conjuntival y el acortamiento del fondo de saco conjuntival, se reduce el ángulo que forma la conjuntiva desde el párpado al fórnix, y se utilizan las letras para describir el grado de acortamiento del fórnix: a) 0-25%, b) 25-50%, c) 50-75% y d) 75-100%.
Estadio 3: se le suman bandas de tejido conectivo que dan lugar al simbléfaron. También se utilizan las letras para determinar el porcentaje de compromiso horizontal del simbléfaron: a) 0-25%, b) 25-50%, c) 50-75% y d) 75-100%. Con la retracción conjuntival progresiva aparece triquiasis, distriquiasis y disminución de la producción de lágrimas.
Estadio final o 4: aparece un síndrome de ojo seco severo, queratinización de la superficie ocular y anquilobléfaron.
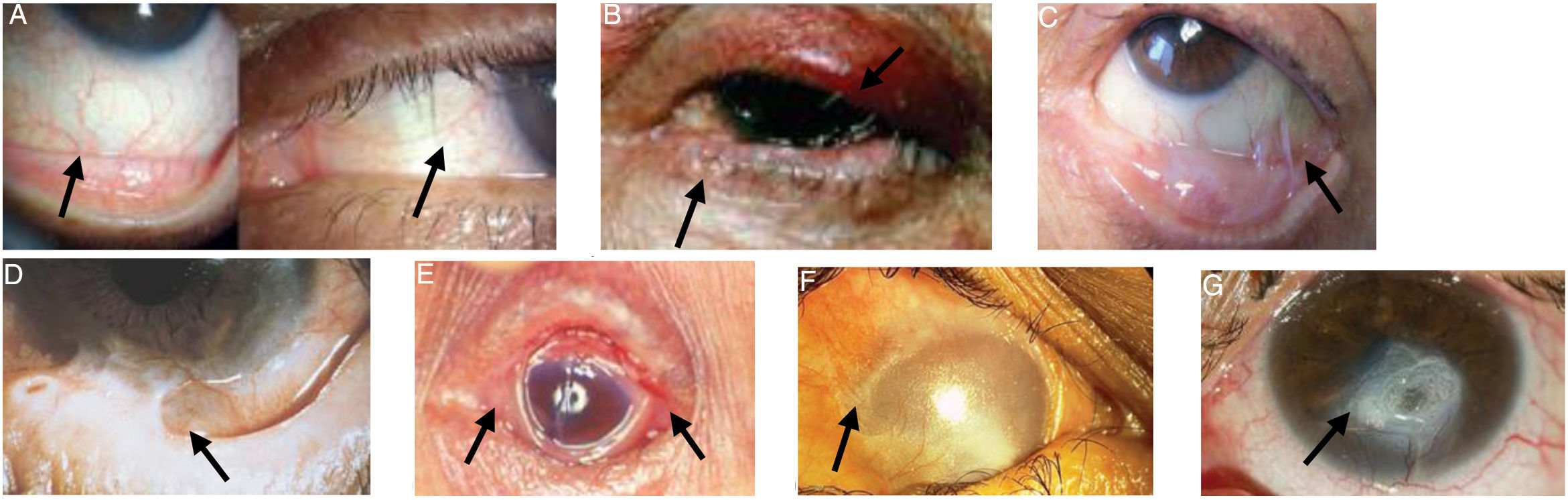
En la figura 1 se presentan imágenes de pacientes con los signos clínicos de los diversos estadios de la enfermedad.
 Enantema conjuntival, estadio 1 (Foster). B) Entropión y distriquiasis. C) Fibrosis subepitelial, acortamiento del fórnix inferior y simbléfaron conjuntival, estadios 2-3 (Foster). D) Simbléfaron más avanzado, estadio 3 (Foster). E) Anquilobléfaron, estadio 4 (Foster). F) Crecimiento epidérmico total sobre superficie ocular. G) Perforación corneal. Figuras adaptadas de López Gamboa et al.75, Espino-Barros Palau et al.12, Chan et al.4, Feizi y Roshandel76, Dart36 y Chirinos-Saldana et al.77.")
Manifestaciones oculares en POC.
A) Enantema conjuntival, estadio 1 (Foster). B) Entropión y distriquiasis. C) Fibrosis subepitelial, acortamiento del fórnix inferior y simbléfaron conjuntival, estadios 2-3 (Foster). D) Simbléfaron más avanzado, estadio 3 (Foster). E) Anquilobléfaron, estadio 4 (Foster). F) Crecimiento epidérmico total sobre superficie ocular. G) Perforación corneal. Figuras adaptadas de López Gamboa et al.75, Espino-Barros Palau et al.12, Chan et al.4, Feizi y Roshandel76, Dart36 y Chirinos-Saldana et al.77.
Una evaluación completa del paciente, con anamnesis y examen físico, ayudará a guiar el diagnóstico. El estándar de oro es la biopsia conjuntival, luego de la cual en la inmunofluorescencia directa (ID) se observa un depósito lineal de Ig o complemento a nivel de la membrana basal conjuntival. La histopatología puede poner de manifiesto otras entidades como, por ejemplo, granulomas en la sarcoidosis o granulomatosis con poliangeítis. La biopsia debe incluir dos muestras de tejido adyacente a un sitio inflamado, una para el estudio anatomopatológico habitual y otra para la ID. En pacientes que presentan compromiso de otras mucosas (PMM), las biopsias iniciales no deben tomarse de la conjuntiva ocular. La muestra obtenida se tiñe con hematoxilina-eosina y con giemsa alcalina. La conjuntiva inflamada muestra infiltración de neutrófilos, macrófagos, células plasmáticas, linfocitos, eosinófilos y células de Langerhans. En etapas avanzadas de la enfermedad, es notable la disminución o ausencia de células caliciformes. Sin embargo, estos hallazgos no son patognomónicos de POC y varían de acuerdo con el estadio de la enfermedad y el sector de toma de muestra. El diagnóstico de certeza se realiza con la ID de la biopsia. Para ello, se deben observar depósitos lineales de IgG, IgM e IgA y c3 en la membrana basal. La positividad de la biopsia varía según las series entre el 20 y 67%. El uso de inmunoperoxidasa en las biopsias con ID negativas de pacientes sospechosos aumentaron la sensibilidad del 52 al 83%6–10,22,23.
En ocasiones, la biopsia puede no ser concluyente y la ID ser negativa. El Grupo para Estudio del Penfigoide Membrano-Mucoso considera que si se cumplen los criterios oftalmológicos de PMM, pero con biopsia no concluyente, se podría definir el caso como PMM con inmunofluorescencia negativa23,24.
Con respecto a los estudios de laboratorio, se han detectado algunos anticuerpos presentes en ciertos casos de PMM, tales como antilaminina 5 y 6, antiantígeno 168kd, anti b4-integrina y antiantígeno de pénfigo bulloso 1 y 2 y anticolágeno tipo VII, entre otros. Es necesario contar con mayor evidencia de su utilidad en los diferentes casos. Por otro lado, no son muy accesibles en la práctica diaria y sus costos son muy elevados. Hasta la fecha, no se dispone de estudios de laboratorio sensibles ni específicos para el diagnóstico y el seguimiento de la respuesta terapéutica del POC7,9,10,18,22–24.
Otros hallazgos oftalmológicos, como sequedad ocular y alteraciones del epitelio corneal, pueden evaluarse mediante diversos test. Entre ellos, el tiempo de rotura lagrimal o tear break-up time (TBUT) es el más utilizado para evaluar la estabilidad de la película lagrimal (tiempo entre un parpadeo completo y la aparición de la primera rotura de la película lagrimal). Se considera anormal un TBUT menor de 10 segundos. Por su parte, el test de Schimer evalúa la producción de lágrimas mediante la colocación de una tira de papel milimetrado entre la mitad externa del párpado inferior y la conjuntiva bulbar de cada ojo, manteniendo los ojos cerrados por 5 minutos. Los valores anormales propuestos varían entre ≤5mm y ≤10mm. Otras medidas correspondientes a ojo seco son la hiperosmolaridad lagrimal, considerada anormal por encima de 308mOsm/L. Para valorar defectos del epitelio corneal se pueden utilizar las tinciones de fluoresceína y su derivado, el verde de lisamina, que tiñen las células dañadas y los filamentos de mucina lagrimal. Finalmente, existen pruebas analíticas como la citología de impresión y la detección de mediadores inflamatorios como citoquinas, metaloproteinasas y factor de necrosis tumoral en las lágrimas25,26.
Dentro de los diagnósticos diferenciales de conjuntivitis cicatrizal (el POC corresponde al 61% de las conjuntivitis cicatrizales) se encuentran las posteriores a quemaduras químicas, procedimientos quirúrgicos oculares, traumas, síndrome de Stevens-Johnson, queratoconjuntivitis epidémica por adenovirus, estreptococo betahemolítico, tracoma, difteria, dermatitis herpetiforme, queratoconjuntivitis atópicas y medicamentosas (fármacos de uso en glaucoma), rosácea ocular, pénfigo vulgar, entre otras. Existen patologías reumatológicas que pueden manifestarse como conjuntivitis cicatrizal, tal es el caso de la granulomatosis con poliangeítis, el síndrome de Sjögren, la esclerodermia, el liquen plano y la sarcoidosis. Se ha reportado ocasionalmente asociado a otras enfermedades autoinmunes, como lupus eritematoso sistémico, artritis reumatoidea, espondilitis anquilosante y tiroiditis autoinmune, entre otras.27–29. El POC también puede ser una manifestación paraneoplásica30,31. Por otro lado, algunos fármacos tópicos y sistémicos pueden causar la aparición de seudopenfigoides, como consecuencia de la exposición de antígenos tras un daño tisular. Entre tales fármacos se citan la pilocarpina, la epinefrina y el ecotiofato tópicos, así como el practolol sistémico9,18,22,32–34.
La media de retraso diagnóstico se ha reportado de 2 años y medio, con una variación de hasta más de 10 años18,22.
TratamientoEl POC es una enfermedad sistémica, por lo que requiere tratamiento local y sistémico de acuerdo con su severidad y evolución. El objetivo está dirigido a controlar la inflamación, facilitar la reparación y prevenir la cicatrización. Dentro del espectro de penfigoides mucosos, aquellos con compromiso ocular se consideran severos y de alto riesgo (al igual que el traqueal o el genital, entre otros), por lo que requieren tratamiento sistémico en todos los casos7,10,18,20,35.
La terapéutica tópica se utiliza de forma adicional al tratamiento sistémico. Los esquemas de formulación se adaptan a las necesidades del paciente y al juicio clínico del oftalmólogo. En el consenso español para las enfermedades de ojo seco, se recomiendan las modificaciones en las condiciones ambientales, dieta y fármacos que podrían alterar la humedad ocular, junto al uso de sustitutos lagrimales de base acuosa o lipídica. En ocasiones, los sustitutos lagrimales son biológicos, como es el caso del suero autólogo y el plasma rico en factores de crecimiento. Pueden adicionarse tratamientos tópicos antiinflamatorios e inmunomoduladores, tales como corticoides, antiinflamatorios no esteroideos, fenilefrina, formulaciones tópicas de ciclosporina A y tacrolimus. También se utilizan estimulantes lagrimales como dicuafosol sódico, rebamipida y N-acetilcisteína3,15,25,26,32.
Se puede recurrir a la oclusión de puntos lagrimales con dispositivos o cauterizándolos si tales dispositivos no son tolerados, de manera que se retengan las lágrimas y se genere alivio. La distriquiasis puede causar queratitis punteada, úlceras y complicar con sobreinfección bacteriana. La depilación se recomienda cada 2 o 3 semanas. Se pueden utilizar lentes de contacto terapéuticos o, en casos avanzados, recurrir a tratamientos quirúrgicos. Sin embargo, dichas intervenciones pueden generar más inflamación y progresión del cuadro. La bléfaro-conjuntivitis es frecuente por la colonización microbiana de la conjuntiva y los bordes palpebrales, de modo que se estimula una mayor inflamación y queratitis. Se ha encontrado una prevalencia de estafilococos en pacientes con penfigoide. Es aconsejable realizar una higiene palpebral diaria y tratar las disfunciones de las glándulas de Meibomio, utilizando antibióticos locales y orales cuando el cuadro lo requiera. De sospecharse sobreinfección, se debe incorporar tratamiento antibiótico3,15,25,26,32. En la tabla 2 se detallan los tratamientos tópicos.
Tratamiento tópico
| Opciones de tratamiento según función | Tipos |
|---|---|
| Sustitutos lagrimales | Lágrimas artificiales de base acuosa o de base lipídica |
| Estimulación lagrimala | Dicuafosol sódico y rebamipidaN-acetilcisteína |
| Sustitutos lagrimales biológicos | Suero autólogo y plasma rico en factores de crecimiento |
| Terapia antiinflamatoria e inmunomoduladora de uso tópico | Corticoesteroides tópicos: hidrocortisona, prednisona, metilprednisolona, dexametasona, fluorometolonaCiclosporina ATacrolimusAntiinflamatorios no esteroideosFenilefrina |
| Antibióticos tópicos | Tetraciclina y análogos (minociclina y doxiciclina)Macrólidos como azitromicina |
| Agentes regeneradores tópicos | Polisulfato de carboximetilglucosaÁcido retinoico (vitamina A o tretionina) |
La dosificación suele variar de acuerdo con la necesidad del paciente y el criterio del oftalmólogo.
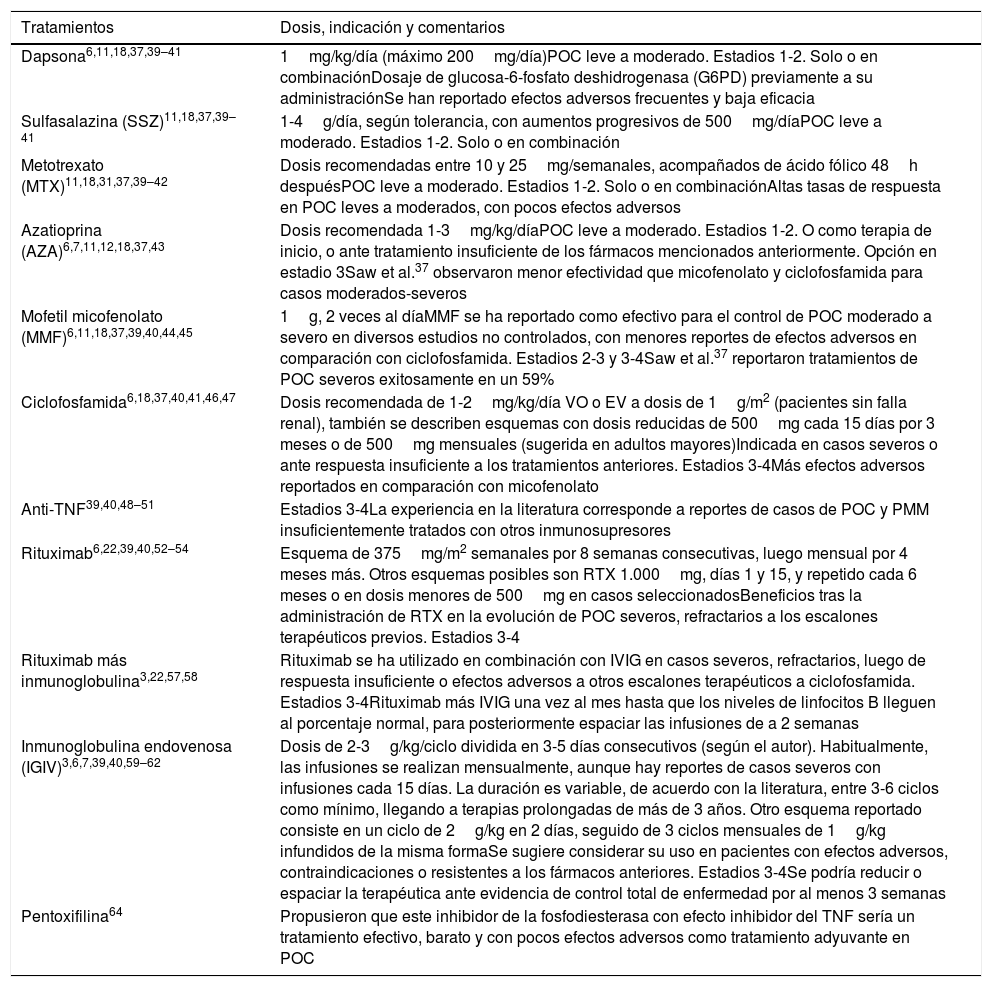
El tratamiento sistémico inmunosupresor se ha establecido clásicamente, de acuerdo con la severidad y de manera escalonada3,7,8,13,36,37. Los cambios o adiciones terapéuticas se han propuesto cada 3 meses si persiste la actividad inflamatoria36,38. Así, en POC leve a moderado (estadios 1-2) se sugiere iniciar con tratamiento tópico más dapsona11,18,37,39–41, sulfasalazina (SSZ)11,18,37,39–41 o metotrexato (MTX)11,18,31,37,39–42, y posteriormente considerar azatioprina (AZA)7,11,18,37,43 o mofetil micofenolato (MMF)11,18,37,39,40,44,45 para casos moderados o moderados severos (estadios 2 o 2-3). Pueden utilizarse combinados (dapsona y SSZ en combinación con MMF, MTX, AZA o ciclofosfamida18,37, o MTX más AZA7). En casos severos (estadios 3-4), MMF o ciclofosfamida18,37,40,41,44,46,47, y en casos muy severos, resistentes (recalcitrantes) o ante efectos adversos, anti-TNF (etanercept e infliximab)40,48–51, rituximab39,40,52–56 o IGIV7,22,39,40,57–63. Otros tratamientos reportados son la pentoxifilina64, la hidroxicloroquina65, el baricitinib y la ciclosporina A66–69 en POC y en PMM. Además de los estadios, es importante considerar en cada caso la actividad de la enfermedad y las características clínicas del paciente7. En la tabla 3 se describen los tratamientos sistémicos y la evidencia disponible de acuerdo con la estrategia de búsqueda, según diseño de estudio.
Tratamientos sistémicos
| Tratamientos | Dosis, indicación y comentarios |
|---|---|
| Dapsona6,11,18,37,39–41 | 1mg/kg/día (máximo 200mg/día)POC leve a moderado. Estadios 1-2. Solo o en combinaciónDosaje de glucosa-6-fosfato deshidrogenasa (G6PD) previamente a su administraciónSe han reportado efectos adversos frecuentes y baja eficacia |
| Sulfasalazina (SSZ)11,18,37,39–41 | 1-4g/día, según tolerancia, con aumentos progresivos de 500mg/díaPOC leve a moderado. Estadios 1-2. Solo o en combinación |
| Metotrexato (MTX)11,18,31,37,39–42 | Dosis recomendadas entre 10 y 25mg/semanales, acompañados de ácido fólico 48h despuésPOC leve a moderado. Estadios 1-2. Solo o en combinaciónAltas tasas de respuesta en POC leves a moderados, con pocos efectos adversos |
| Azatioprina (AZA)6,7,11,12,18,37,43 | Dosis recomendada 1-3mg/kg/díaPOC leve a moderado. Estadios 1-2. O como terapia de inicio, o ante tratamiento insuficiente de los fármacos mencionados anteriormente. Opción en estadio 3Saw et al.37 observaron menor efectividad que micofenolato y ciclofosfamida para casos moderados-severos |
| Mofetil micofenolato (MMF)6,11,18,37,39,40,44,45 | 1g, 2 veces al díaMMF se ha reportado como efectivo para el control de POC moderado a severo en diversos estudios no controlados, con menores reportes de efectos adversos en comparación con ciclofosfamida. Estadios 2-3 y 3-4Saw et al.37 reportaron tratamientos de POC severos exitosamente en un 59% |
| Ciclofosfamida6,18,37,40,41,46,47 | Dosis recomendada de 1-2mg/kg/día VO o EV a dosis de 1g/m2 (pacientes sin falla renal), también se describen esquemas con dosis reducidas de 500mg cada 15 días por 3 meses o de 500mg mensuales (sugerida en adultos mayores)Indicada en casos severos o ante respuesta insuficiente a los tratamientos anteriores. Estadios 3-4Más efectos adversos reportados en comparación con micofenolato |
| Anti-TNF39,40,48–51 | Estadios 3-4La experiencia en la literatura corresponde a reportes de casos de POC y PMM insuficientemente tratados con otros inmunosupresores |
| Rituximab6,22,39,40,52–54 | Esquema de 375mg/m2 semanales por 8 semanas consecutivas, luego mensual por 4 meses más. Otros esquemas posibles son RTX 1.000mg, días 1 y 15, y repetido cada 6 meses o en dosis menores de 500mg en casos seleccionadosBeneficios tras la administración de RTX en la evolución de POC severos, refractarios a los escalones terapéuticos previos. Estadios 3-4 |
| Rituximab más inmunoglobulina3,22,57,58 | Rituximab se ha utilizado en combinación con IVIG en casos severos, refractarios, luego de respuesta insuficiente o efectos adversos a otros escalones terapéuticos a ciclofosfamida. Estadios 3-4Rituximab más IVIG una vez al mes hasta que los niveles de linfocitos B lleguen al porcentaje normal, para posteriormente espaciar las infusiones de a 2 semanas |
| Inmunoglobulina endovenosa (IGIV)3,6,7,39,40,59–62 | Dosis de 2-3g/kg/ciclo dividida en 3-5 días consecutivos (según el autor). Habitualmente, las infusiones se realizan mensualmente, aunque hay reportes de casos severos con infusiones cada 15 días. La duración es variable, de acuerdo con la literatura, entre 3-6 ciclos como mínimo, llegando a terapias prolongadas de más de 3 años. Otro esquema reportado consiste en un ciclo de 2g/kg en 2 días, seguido de 3 ciclos mensuales de 1g/kg infundidos de la misma formaSe sugiere considerar su uso en pacientes con efectos adversos, contraindicaciones o resistentes a los fármacos anteriores. Estadios 3-4Se podría reducir o espaciar la terapéutica ante evidencia de control total de enfermedad por al menos 3 semanas |
| Pentoxifilina64 | Propusieron que este inhibidor de la fosfodiesterasa con efecto inhibidor del TNF sería un tratamiento efectivo, barato y con pocos efectos adversos como tratamiento adyuvante en POC |
En cuanto a la utilización de corticoides, esta se reserva para el control agudo de la enfermedad y mientras se aguarda el efecto completo de los demás fármacos inmunosupresores. Las dosis y la vía de administración se basan en la severidad del caso. Si la actividad es moderada-severa, se debe considerar prednisona 1mg/kg/día o pulsos de metilprednisolona 500mg/día por 3 días, con posterior descenso paulatino de la dosis. En casos leves o leves-moderados, dosis variables entre 5-40mg de prednisona podrían ser suficientes para acompañar temporalmente a los demás fármacos inmunosupresores. Se sugiere utilizar la mínima dosis y por el menor tiempo posible que logre controlar los síntomas7,36,37,40.
El tratamiento inmunosupresor debe ser continuado por al menos un año después de la estabilización de la enfermedad, y posteriormente se debe considerar un descenso escalonado7,13,18,36,37,39,40. Saw et al.37 observaron que un 41% de los ojos evaluados (92/223) presentaron progresión de la cicatrización, continuando con actividad inflamatoria mínima o leve, no posible de eliminar a pesar de la inmunosupresión, o tras la discontinuación del tratamiento. Otros reportaron cicatrización aun en ausencia de actividad inflamatoria detectable18,36. Estos hallazgos sugieren que el proceso cicatrizal puede continuar a pesar del control de la actividad inflamatoria visible clínicamente y que los tratamientos inmunosupresores actuales pueden no llegar al control completo de la patología. Uno de cada 3 pacientes recae en el transcurso de los años posteriores a la disminución o suspensión de la inmunosupresión, por lo que es necesario un seguimiento estrecho a pesar de la remisión18,36,37,39,40,68.
Las respuestas a los tratamientos se clasifican de la siguiente manera: exitoso, cuando la enfermedad se mantiene quiescente, con ojo blanco por al menos 3 meses desde el inicio de la inmunosupresión; parcialmente exitoso, cuando hay control parcial de la inflamación, pero persiste inflamación residual; falla, ante respuesta insuficiente al fármaco, continuando con inflamación o ante suspensión del medicamento por efectos adversos37. Otras definiciones en la literatura corresponden a: remisión, ante la ausencia de cicatrización progresiva y de actividad ocular inflamatoria por ≥2meses; remisión parcial, cuando hay mejoría clínica y control de la enfermedad por ≥2meses; y remisión a largo plazo, cuando ante la suspensión del tratamiento inmunosupresor por ≥1año, no hay signos clínicos detectables de progresión de acuerdo con los estadios de Foster70,71.
DiscusiónEn esta revisión narrativa, la evidencia sobre la clínica de presentación es abundante, con manifestaciones variadas. En relación con el diagnóstico, hay acuerdo en que el estándar de oro es la biopsia, pero si esta es negativa o se carece de inmunofluorescencia y la clínica es característica, también se puede sospechar el diagnóstico. Se destaca la demora en el diagnóstico y la clínica insidiosa y potencialmente atribuible a otras patologías más frecuentes al inicio de la enfermedad, generalmente con conjuntivitis a repetición, ojo rojo, sequedad ocular, entre otros. Sin embargo, la evidencia en la literatura jerarquiza la necesidad de un diagnóstico y tratamiento oportuno para prevenir secuelas oftalmológicas.
En los últimos 20 años, la evidencia sobre el tratamiento sistémico proviene en su mayoría de estudios observacionales, con algunos experimentales. Sin embargo, se destacan ensayos clínicos aleatorizados realizados previamente a la fecha de búsqueda que demostraron la eficacia del uso de ciclofosfamida y prednisona en estadios avanzados de la enfermedad1,72,73, además de una revisión Cochrane sobre intervenciones en penfigoide mucocutáneo y epidermólisis bullosa74. No se han encontrado metaanálisis según la estrategia de búsqueda, solo se encontró un estudio clínico aleatorizado para pentoxifilina. De acuerdo con los diseños de estudio, la literatura presentada corresponde en su mayoría a calidad II-III. Por otro lado, se observa una mayor cantidad de estudios en relación con rituximab e inmunoglobulina en comparación con otros utilizados en estadios previos.
En la actualidad los tratamientos se basan en la estrategia escalonada, pero la progresión con cicatrización irreversible y la presencia de inflamación blanca cuestionan si este abordaje es correcto en todos los casos, o si una mayor inmunosupresión debería ser iniciada precozmente en ciertos pacientes para prevenir la evolución a fibrosis. Aún no hay suficiente evidencia para predecir qué pacientes desarrollarán un curso agresivo de la enfermedad y si se beneficiarían de un tratamiento más intensivo desde el inicio, principalmente en aquellos cuya única manifestación es POC sin compromiso extraocular. En algunas oportunidades, las recomendaciones se extrapolan de la evidencia en el tratamiento de MMP, otras patologías oculares como uveítis, u otras enfermedades autoinmunes sistémicas, por lo que es necesario ampliar el conocimiento farmacocinético y farmacodinámico a nivel oftalmológico. Asimismo, es probable que en la literatura se encuentren predominantemente casos con evoluciones favorables y, por lo tanto, un subregistro de aquellos con desenlaces negativos. Existe una amplia variedad de tratamientos cuyas indicaciones varían de acuerdo con los hallazgos oftalmológicos, sin embargo, todavía no hay suficiente evidencia en cuanto a la mejor recomendación en cada caso.
Debido a que el comienzo del cuadro suele ser inespecífico, los pacientes tienden a deambular por distintas instituciones y profesionales, consultando con frecuencia a guardias solo ante aumentos de la sintomatología. El tiempo generalmente acotado de estas consultas, así como la imposibilidad de seguimiento por un mismo profesional, conducen a un mal seguimiento del cuadro. A su vez, la dificultad para la realización de la biopsia conjuntival (costos elevados, falta de cobertura por algunos sistemas de salud, necesidad de patólogos especializados en el tema, etc.) hace que el diagnóstico y el tratamiento se retrasen. En casos severos, diagnosticados y tratados tardíamente, la progresión de la enfermedad puede llevar a secuelas y pérdida de la visión, por lo que el gran desafío médico es el diagnóstico precoz y su correspondiente tratamiento.
ConclusiónEl POC es una patología poco frecuente en comparación con otras enfermedades autoinmunes, de severidad variable, en su mayoría casos leves a moderados. La presentación clínica muchas veces inespecífica y las dificultades en la realización de la biopsia conjuntival tienen como consecuencia un retraso en el diagnóstico y en la inmunosupresión, lo cual resulta en la progresión de la enfermedad con fibrosis. La evidencia disponible sobre los tratamientos inmunosupresores, principalmente en casos severos o refractarios a las terapéuticas iniciales, son limitados, con estudios en su mayoría no controlados, un número reducido de pacientes y seguimientos cortos.
La determinación de factores de riesgo para la progresión a enfermedad cicatrizal severa y de marcadores serológicos o en biopsias conjuntivales constituye un desafío hacia el futuro, a fin de predecir qué pacientes van a desarrollar una enfermedad de progresión rápida y agresiva y cuáles van a tener una evolución lenta y muy raramente desarrollen una enfermedad mucosinequiante.
En las últimas décadas el ser humano ha aumentado su expectativa de vida, lo cual hace que conviva con enfermedades crónicas. Un alto índice de sospecha y conocimiento sobre el tema es necesario para evitar secuelas oftalmológicas definitivas y mejorar la calidad de vida de los pacientes. Se destaca el abordaje interdisciplinario entre oftalmólogos, reumatólogos e inmunólogos. Los hallazgos de la presente revisión narrativa exploran la evidencia disponible sobre las características clínicas, diagnósticas y terapéuticas, siendo aplicables en su mayoría en la práctica diaria. La validez de los estudios debe ser interpretada con base en la calidad y el diseño de cada uno de ellos. Finalmente, es fundamental continuar el estudio de esta entidad, para mejorar la calidad de la evidencia disponible y de esta forma optimizar los abordajes diagnósticos y terapéuticos.
Conflicto de interesesNinguno.
FinanciaciónNinguna.
