



Pachymeningitis is an uncommon disease with multiple etiologies with fibrosis of leptomeninges. One type of this disease is related to immunoglobulin G4 (IgG4). The most frequent manifestations include chronic cephalgia and focal neurological deficits. The case of a 31-year-old man with chronic cephalgia localized to the left hemicranium and left dysmetria is presented. Nuclear magnetic resonance imaging showed image hyperintensity and perilesional edema located at the infratentorial and left parietal occipital levels. The results of cerebral angiography and a cerebrospinal fluid examination were normal. A biopsy of the central nervous system (CNS) ruled out neoplastic lesions, granulomatous lesions, infection, and arteriovenous malformation and indicated thickened meningeal fragments. Elevated serum IgG4 levels were found. The antinuclear antibodies (ANAs), antibodies to extractable nuclear antigens (ENAs), anti-native DNA antibodies (anti-DNA), topoisomerase I antibodies (Scl-70), antibodies to cyclic citrullinated peptides (anti-CCP), and rheumatoid factor (RF) were negative. After an initial diagnosis of idiopathic hypertrophic cranial pachymeningitis, the patient received treatment with high doses of glucocorticosteroids for two years, without response, and developed Cushing's syndrome, cataracts, and osteoporosis. During the follow-up, dactylitis, distal upper limb sclerosis, and Raynaud's phenomenon occurred. A capillaroscopy indicated capillaries with dilated loops, microhemorrhaging, disorganization of the capillary architecture, and megacapillaries, which led to a diagnosis of limited systemic scleroderma. Although an association between scleroderma and pachymeningitis was originally proposed, review of the literature suggests that these are two different entities. Treatment with methotrexate, 25mg weekly, allowed the withdrawal of glucocorticoids and resolution of neurological symptoms.
La paquimeningitis es el compromiso fibrosante de las leptomeninges poco común y de múltiple etiología. Una de ellas, la enfermedad relacionada a inmunoglobulina IgG4. Las manifestaciones clínicas más frecuentes son la cefalea crónica de difícil tratamiento y el déficit neurológico focal. Se presenta el caso de un hombre de 31 años, con cefalea crónica localizada en el hemicráneo izquierdo y dismetría izquierda, resonancia nuclear magnética con imagen hiperintensa y edema perilesional ubicada a nivel infratentorial y parieto-occipital izquierda, angiografía cerebral y estudio de líquido cefalorraquídeo normales. La biopsia del sistema nervioso central (SNC) descartó lesiones neoplásicas, granulomatosas, infección y malformación arteriovenosa, y reportó fragmentos meníngeos engrosados. A nivel sérico se encontraron altos niéveles de IgG4. ANA, ENA, anti-DNA, SCL-70, CCp y FR, negativos. Con el diagnóstico presuntivo inicial de paquimeningitis craneal hipertrófica idiopática, recibió tratamiento con dosis elevadas de glucocorticoides por espacio de 2 años, sin buena respuesta y el desarrollo de síndrome de Cushing iatrogénico, cataratas y osteoporosis. Durante el seguimiento se documentó dactilitis, esclerosis distal de miembros superiores, síndrome de Raynaud y con capilaroscopia con asas dilatadas, microhemorragia, desorganización de la arquitectura capilar y megacapilares, configurándose el diagnóstico de esclerodermia sistémica limitada. Aunque se propuso inicialmente la asociación entre estas 2 condiciones, la revisión de la literatura indica que se trata de 2 entidades fisiopatológicamente diferentes. El tratamiento con metotrexato a dosis de 25mg semanal permitió el retiro de los glucocorticoides y la resolución completa de los síntomas neurológicos.
IgG4-related disease is a newly recognized clinicopathologic entity characterized by the development of space-occupying fibrosclerosing lesions that cause tissue damage and organ dysfunction in multiple potential targets. Although it is not recognized under the currently established nomenclature, the first descriptions of this entity were recorded in patients with a unique type of chronic autoimmune pancreatitis. Subsequent reports have documented the presence of similar histopathological findings in other organs and structures such as the thyroid gland, salivary glands, lacrimal glands, kidney, periaortic space, mediastinum, and retroperitoneum, among others. Recently, was described the compromise of meninges and pituitary gland, establishing the central nervous system as another potential target of this systemic disease. Over time, experts on the subject have established a set of diagnostic criteria that will allow future inclusion of select population groups, which is essential for the execution of controlled clinical trials and has been absent up to this point. Based on these diagnostic criteria, a possible case of IgG4-related meningeal disease is presented, and its therapeutic approach and the appropriate evolution with the established behavior are also discussed.
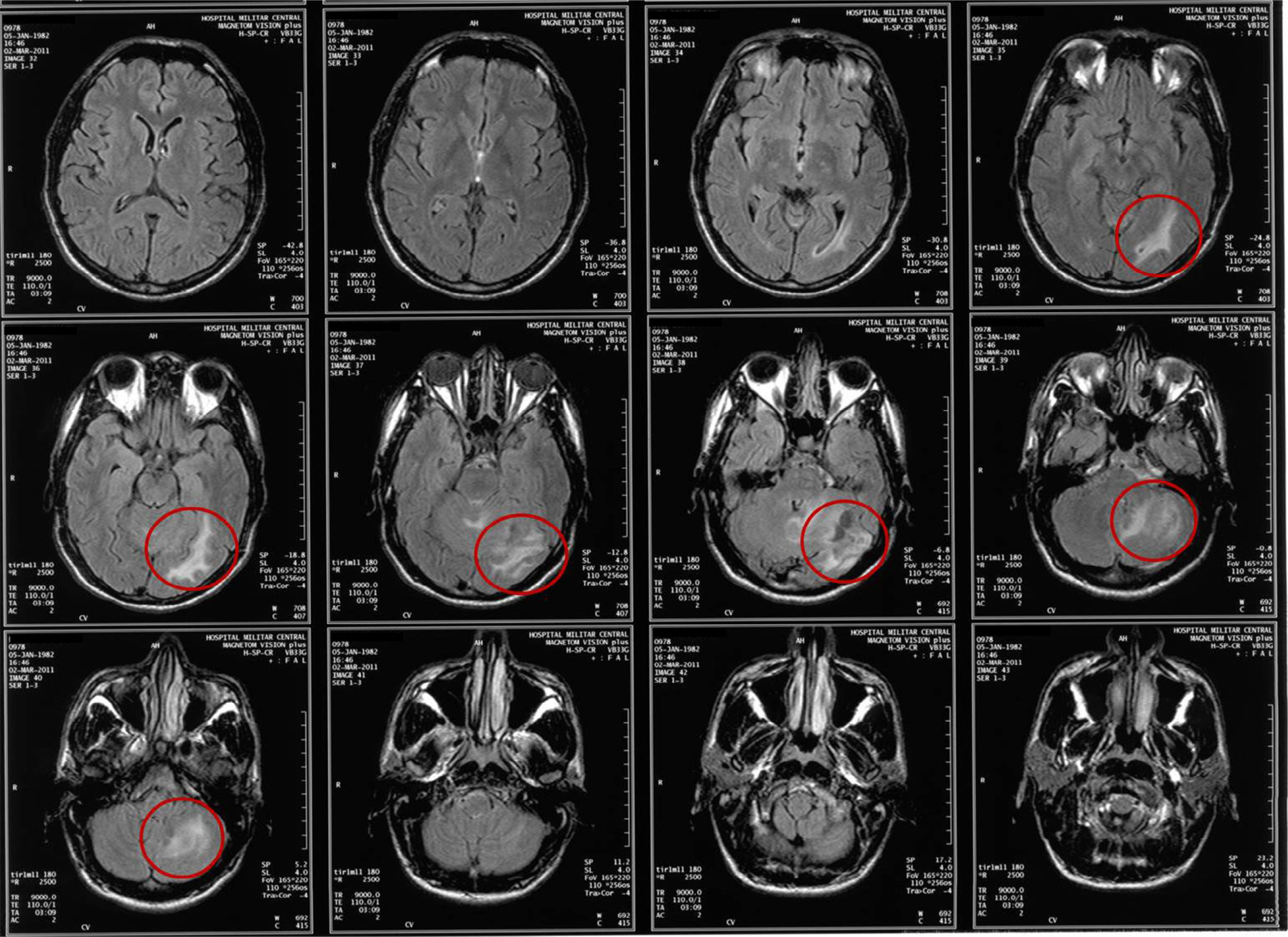
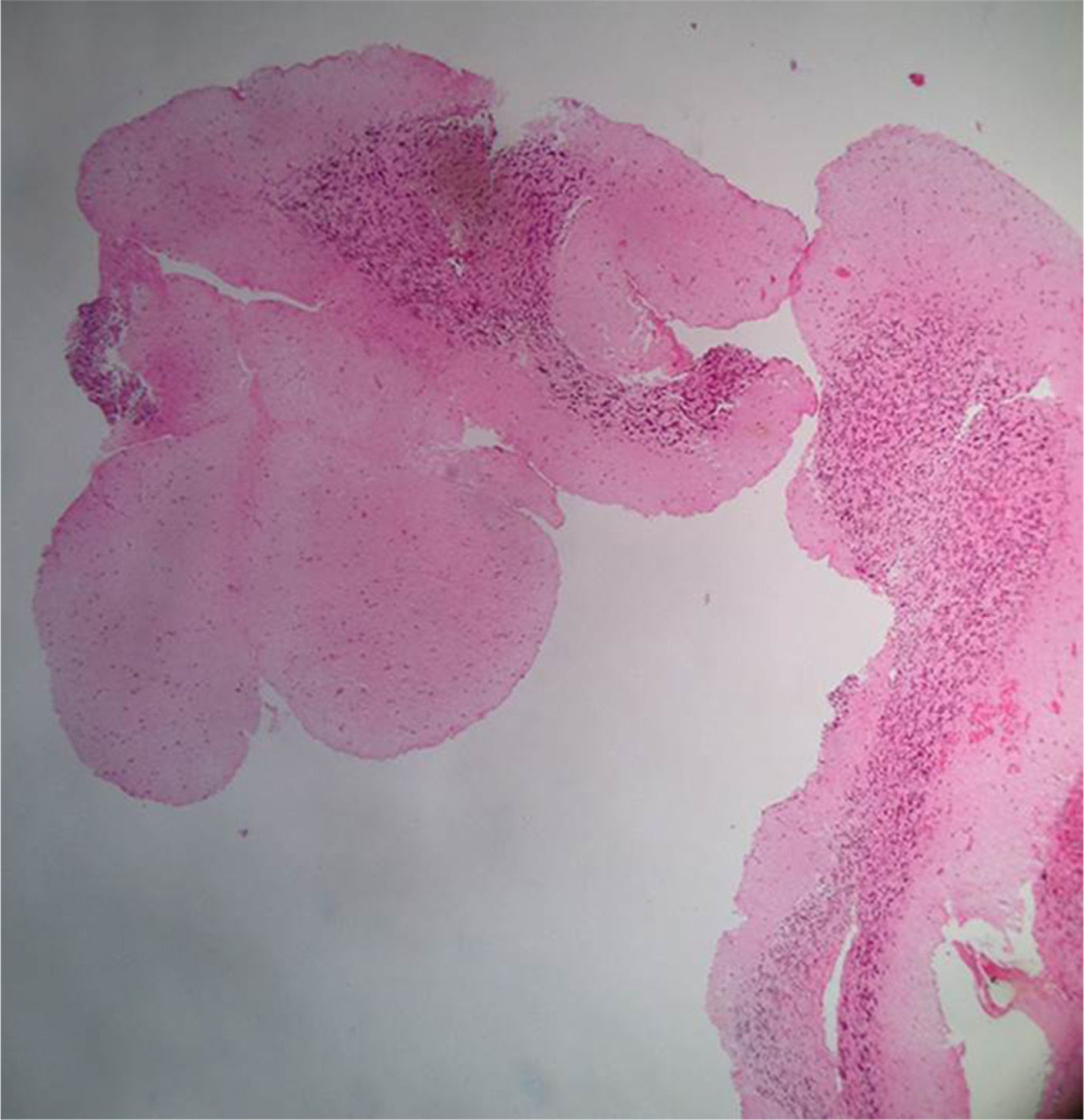
Clinical caseA 31-year-old man consulted the emergency department for a simple partial seizure episode that generalized into tonic–clonic seizures. The patient, after a year of evolution, presented severe global cephalgia associated with two previous convulsive episodes with similar characteristics. He had a history of a resolved depressive disorder. He was receiving treatment with phenytoin (300mg orally) every day. Upon admission to the emergency department, he exhibited a postictal state, no focal neurological deficits, a BP of 149/80mm Hg, a heart rate of 96 per minute, a respiratory rate of 21 per minute, and a body temperature of 37.1°C. At the time, he was considered to be coursing with cephalgia symptomatic of a long-term evolution of focal neurological symptoms. A cranial nuclear magnetic resonance imaging examination showed image hyperintensity with evidence of the presence of perilesional edema located at the infratentorial and the left parietal occipital levels (Fig. 1). After recovery of consciousness, a detailed neurological examination showed mild left dysmetria, preserved higher mental functions, normal strength and muscle tone, no meningeal signs, cranial nerves without alterations, and normal nociception and proprioception. A diagnostic and therapeutic craniotomy was performed. A poorly vascularized, solid, yellowish lesion that adhered to the tentorium and cerebellum was found; the arachnoid membrane was thickened and whitish. A pathology examination revealed the presence of multiple normal cerebellar cortex fragments and thickened meninges. No findings consistent with vasculitis, infections, granulomatosis, malignancy, metastases, or arterial or venous malformations were found (Fig. 2). A cerebral pan-angiography examination was normal. A cerebrospinal fluid examination was normal. Cultures for common microbes, fungi, and mycobacteria were negative. The VDRL, ADA and PCR assays for Mycobacterium tuberculosis and the latex agglutination test for Cryptococcus were negative. The antinuclear antibodies (ANAs) were positive at a 1/160 DIL with a centromere pattern. The anti-native DNA antibodies (anti-DNA), antibodies to extractable nuclear antigens (ENAs), topoisomerase I antibodies (Scl-70), rheumatoid factor (RF), and antibodies to cyclic citrullinated peptides (anti-CCP) were negative. Based on these findings, the Neurology and Neuroradiology Department made the diagnosis of idiopathic hypertrophic cranial pachymeningitis, and treatment was initiated with prednisolone (1mg/kg/day). The patient received treatment with high doses of oral glucocorticoids for the following two years and showed partial remission with exacerbation of episodes of cephalgia, which did not allow a reduction in the dose of prednisolone. The patient developed iatrogenic Cushing's syndrome, cataracts, and glucocorticoid-induced osteoporosis, conditions for which he was referred to the Rheumatology Department.


Pathology report, April 20, 2011. Microscopic description: Material represented by free fragments of cerebellar cortex. Three layers on meneinges are identified. There on the subarachnoid space arterial wall fragments without transition to venous wall. In the material examined are not observed nonneoplastic or arteriovenous malformation or metastatic lesion.
During the evaluation by the Rheumatology Department, distal skin thickening of the hands and dactylitis were found. A capillaroscopy examination showed dilated capillary loops, microhemorrhages, disorganization of the capillary architecture, and megacapillaries. Under the presumptive diagnosis of limited systemic scleroderma, the following evaluations were performed: a spirometry and volume flow curve was generated, which showed results consistent with a mild restrictive pattern, and esophagogastroduodenoscopy and echocardiogram results were within normal limits. Parenteral treatment with methotrexate was initiated at a dose of 25mg weekly. After six months of treatment, the patient presented resolution of symptoms, permitting the progressive reduction and withdrawal of treatment with glucocorticoids. In addition, the patient's serum cholesterol and triglyceride levels normalized, bone mineral density increased, and weight decreased.
DiscussionPachymeningitis is a chronic inflammatory disease that results in fibrosing, which generates a localized or diffuse thickening principally of the dura mater (pachymeninx), but it can also compromise, in isolation or concurrently, the arachnoid mater and the pia mater (leptomeninges). Pachymeningitis was first described in 1869 at the spinal level by Drs. J.M. Charcot and A. Joffroy and at the cranial level in 1949 by Drs. W.E. Stern and H.C. Naffziger.1 It may be of primary or secondary origin and associated with infectious processes (tuberculosis, Lyme disease, cysticercosis, neurosyphilis, human T-lymphotropic virus I, and fungal infection), autoinflammatory or systemic autoimmune processes (sarcoidosis, polyangiitis granulomatosis, temporal arteritis, Sjögren's syndrome, and rheumatoid arthritis), malignant neoplastic processes (meningioma, dural carcinomatosis, and metastasis), and vascular processes (angiitis of the central nervous system).2 Recently, the relationship between the IgG subtype 4 reservoir and the appearance of meningeal sclerosing lesions has been described. The report presented by Dr. Chan et al.3 was the first to establish the possibility that IgG4-related pachymeningitis represented a subgroup of the classically designated idiopathic hypertrophic pachymeningitis. Since then, approximately 33 cases (21 men) of IgG4-related pachymeningitis have been reported in the medical literature, all confirmed by biopsy.4 Despite its first description in 2009, it was not until 2012, at a global meeting of experts in pathology, neurology, diagnostic imaging, rheumatology, and basic sciences, that it was established by mutual agreement that a compromise of the central nervous system (meninges and hypophysis) related to IgG4 was part of a spectrum of potential target organs of a systemic multiorgan sclerosing disease associated with IgG4. At that meeting, the nomenclature for each of the potentially affected organs was defined, and the terms “hypophysitis related to IgG4” and “pachymeningitis related to IgG4” were fixed.5
The first attempt to establish a set of diagnostic criteria for IgG4-related disease resulted from the original work of the Japanese Ministry of Health, Labor, and Welfare, which was led by Drs. H. Umehara and K. Okazaki, who gathered a group of experts on the subject and specified a series of criteria for an integrated diagnostic approach to IgG4-related disease. These criteria are based on clinical, imageological, serological, and immunohistopathological findings. Based on these findings, an algorithm was proposed (Fig. 1) that permits the categorization of the diagnosis as possible, probable, or definitive.6 Although clinical and radiological studies indicate that the findings are different, depending on the organ affected, the clinical diagnosis is conditional on the presence of a swollen mass lesion that determines, to a variable degree, the dysfunction of the target organ.6,7 Regarding the serology of patients with IgG4-related disease, it is important to note that generally (but not always) the acute phase reactants, C-reactive protein, and erythrocyte sedimentation rate are found to be moderately elevated, while it is usual to find negative or weakly positive ANAs in low titers; likewise, the ENAs are most frequently found to be negative. It is noteworthy that this is an entity in which the identification of a specific antibody and antigen as a triggering factor of the disease has not been achieved.4,8 Elevated serum IgG4 levels are found in approximately 70–90% of patients with IgG4-related disease, and it seems that the degree of elevation is greater in the case that multiple organs are compromised than when one organ is affected. Based on the studies of Drs. M. Yamamoto and Y. Masaki of the School of Medicine of Sapporo and of the University of Medicine of Kanazawa, performed on an Asian population in 2012, the operative characteristics of the measure were determined by nephelometry of serum IgG4 levels as a diagnostic tool in IgG4-related disease. According to the study by Dr. M. Yamamoto's group, which was conducted with a sample of 402 patients recruited between October 2010 and March 2011, with the objective of determining the cutoff point of the serum IgG4 level for which the highest diagnostic yield could be obtained for an IgG4-related disease different from type 1 autoimmune pancreatitis (AIP). For an IgG4 value of 144mg/dl (AIP=135mg/dl), a sensitivity of 95.1% and a specificity of 90.7% were obtained, and an area under the ROC curve of 0.97 was determined. In the same year, the team of Dr. Y. Masaki independently conducted a retrospective analysis of 132 patients with IgG4-related disease and 48 controls with different diagnoses and determined that for a cutoff point of serum IgG4>135mg/dl, a sensitivity of 95.5% and a specificity of 87.5% were achieved.9,10 Very recently, while conducting a retrospective analysis of 190 medical records of patients with elevated serum IgG4 levels and 190 patients with normal serum levels recruited between 2001 and 2011, Drs. M. Carruthers and J. Stone of the Division of Immunology and Rheumatology at Massachusetts General Hospital in Boston determined that in an American population, using a cutoff point of serum IgG4>135mg/dl, a sensitivity of 90%, a specificity of 60%, a positive predictive value of 34%, and a negative predictive value of 96% were achieved. This divergence of results could be explained by the heterogeneity of populations (Asian vs. American), the variability of the measuring instruments, and methodological divergence (prospective vs. retrospective). However, there are additional points to be analyzed in these articles, but they are beyond the current intention of this writing.5,10 It now appears that the IgG4 level in the cerebrospinal fluid is more important than the blood level of IgG4.11
As for the immunohistochemistry component, the diagnostic algorithm was based on mutual agreement between specialists in the subject matter in 2012. The characteristic features of the IgG4-related disease were recognized and defined as follows: lymphoplasmacytic infiltration of IgG4-positive cells by immunohistochemistry, radiated fibrosis (cartwheel), and obliterative phlebitis.10 If a patient has two of these three features combined with elevated serum IgG4 levels, the diagnosis is definitive, but the diagnosis is probable if serum IgG4 levels are low or normal and histopathology reveals positive findings. Conversely, if there is a clinical condition and some highly suggestive images with a serum IgG4 level above the cutoff point but the histopathological findings are not positive, the patient is stratified as possible for IgG4-related disease.10,12
Based on the previously mentioned characteristics and to the best of our knowledge, in our country, there have been no formally reported cases of IgG4-related pachymeningitis, but there have been reports of meningeal thickening of an apparent idiopathic nature.13 Using the above explanations as a reference framework, we presented a report of a possible case of IgG4-related pachymeningitis according to the most likely diagnosis once the differential diagnoses were discarded based on the daily clinical presentation frequency. We argue that according to the medical literature, it is not always possible to obtain positive histopathological results because the presence of these results is dependent on the technique of sample collection (fine needle biopsy, excisional biopsy, or cellular block), the stage of evolution (greater likelihood of positive findings in early stages of the disease), and localization of the material collected for study (higher probability of positive findings at the pericapillary level).10 Therefore, it is possible to not obtain a positive immunohistochemical study.
Regarding the treatment of IgG4-related disease, it has been stated from the first descriptions of cases that the cornerstone of management is steroids; no guide to treatment, or at least an outline, has been established that defines an agreed upon initial dose (pulses or high or medium doses), strategy of reduction and suspension of the glucocorticoids, and duration of the therapy. The majority of the reports have demonstrated a satisfactory response achieved in terms of weeks with respect to control of the symptoms, improvement of organic dysfunction, and reduction in size of the mass type lesion; however, a proportion of patients who are non-responders or who develop intolerable adverse effects has been observed. For this group of patients, empirical testing has been performed by using synthetic immunomodulatory drugs such as azathioprine,14 mycophenolate mofetil, and methotrexate (MTX);15–17 biological therapy consisting of B-cell depletants (Rituximab);18 and antineoplastics such as cytarabine.19 The results of these treatments have been divergent. For the use of MTX, reports found in the literature relate a greater experience in the handling of meningeal disease with a suitable profile of efficacy and safety.17 Based on this finding, we used MTX as an immunomodulator and achieved gradual withdrawal of steroids without symptomatic recurrence and with adequate reversal of the side effects.
It is finally advised that, despite being a fibrosclerosing entity, IgG4-related disease is a completely different condition than limited scleroderma, systemic, and related syndrome. The arguments for this difference lie in the divergence between the pathophysiological mechanisms in each entity as well as in a retrospective review published by the group of Dr. D.M. Reddi of the Pathology Department at Duke University,20 who performed an immunohistochemical assessment of plates taken from the archives of surgical pathology specimens or autopsies, both with confirmed clinical diagnoses of scleroderma or related syndromes (cases) and normal skin biopsies (controls). They assessed the absolute number of IgG4-positive plasma cells by high-power field (HPF) microscopy and the ratio of IgG4-positive/IgG-positive plasma cells in the cases and controls and determined that counting more than 10 cells/HPF and a cutoff point>30% for the ratio of IgG4/IgG plasma cells were conclusive of IgG4-related disease. Of all the cases assessed with scleroderma and related syndromes, only one case exhibited positive immunohistochemistry exceeding the >30% cutoff point. Based on this information, it was concluded that no relationship exists between scleroderma and IgG4-related disease, despite the fibrosclerosing nature of the two. The work of Dr. D.M. Reddi contradicts some editorials that have attempted to imply some relationship between these two conditions.21
Ethical disclosuresProtection of human and animals subjectsThe authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data.
Right to privacy and informed consentThe authors must have obtained the informed consent of the patients and/or subjects mentioned in the article.
Conflicts of interestThe authors declare that they have no conflict of interest.
