





Psoriasis is a disease that is not limited to skin involvement, the importance of systemic compromise is recognized more than ever, especially because of the associated comorbidities, which are explained by the marked chronic systemic inflammatory response mediated by pro-inflammatory cytokines (mainly TNF-α, IL23, IL 17), which play an important role in the induction of insulin resistance, endothelial dysfunction, accelerated atherosclerosis and the increased risk of cardio-cerebrovascular events. The relationship with these outcomes has been demonstrated and hence the concept of psoriatic march, a term that is gaining increasing importance aimed at maintaining and reinforcing the approach to a skin disease with systemic compromise, associated morbidity and mortality that can be preventable and manageable. The pathophysiological mechanisms that explain these phenomena are variable, however, new concepts have been identified, which have made it possible to improve the current approach to the disease and thus establish mechanisms to reduce cardiovascular risk in patients with psoriasis.
La psoriasis es una enfermedad que va más allá de la afectación exclusivamente cutánea, cada día se reconoce más la importancia del compromiso sistémico, en especial por las comorbilidades asociadas. Dichas comorbilidades se explican por la marcada respuesta inflamatoria sistémica crónica mediada por citoquinas proinflamatorias (principalmente TNF-α, IL-23, IL-17), las cuales desempeñan un papel importante en la inducción de la resistencia a la insulina, la disfunción endotelial, la aterosclerosis acelerada y el aumento del riesgo de eventos cardio-cerebrovasculares. La relación con estos desenlaces ha sido demostrada y es allí donde surge el concepto de marcha psoriásica, un término que cobra cada día más importancia, cuyo objetivo es mantener y reforzar el enfoque de una enfermedad cutánea con compromiso sistémico, morbimortalidad asociada que puede ser prevenible y manejable. Los mecanismos fisiopatológicos que explican estos fenómenos son variables, sin embargo, se han dilucidado nuevos conceptos que han permitido mejorar el enfoque actual de la enfermedad y así establecer mecanismos para disminuir el riesgo cardiovascular en pacientes con psoriasis.
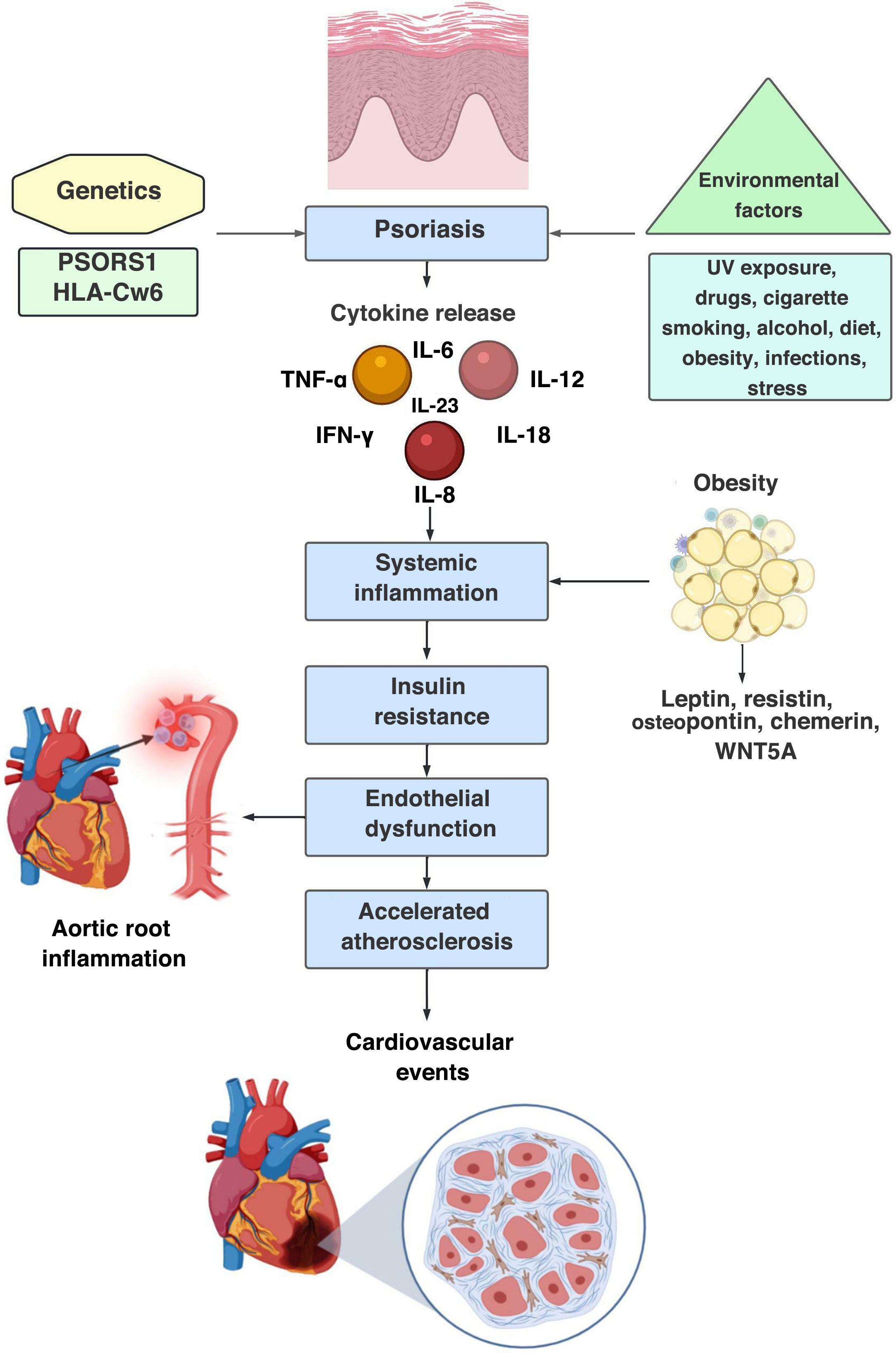
Psoriasis (Pso) is a chronic systemic disease, mediated by immune mechanisms, environmental factors1 and genetic susceptibility, in which the PSORS1 gene stands out as the main genetic determinant and HLA Cw6 as a susceptibility factor for the early onset of the disease.2,3 (Table 1) Its global prevalence ranges around 2%–3%, with no differences in distribution by gender, while its incidence has a bimodal peak, the first between 18 and 39 years of age and the second between 50 and 69 years of age.2 It is characterized by heterogeneous cutaneous involvement and the most frequent lesions are well defined erythematous plaques, with silvery-gray scales, while other forms include papules and drop-shaped plaques, and pustules, among others, with a variable degree of extension, that can be located anywhere in the body.1 Imaging techniques such as magnetic resonance and pulsed-Doppler musculoskeletal ultrasound have allowed to document sites of inflammation beyond the skin,4 among them the enthesis organ (the site of insertion to the bone of tendons, ligaments, articular capsule, annulus fibrous of the intervertebral disk), which is the initial site of inflammation in psoriatic arthritis (Psa),5 that can occur in up to 30% of the patients with Pso.3 Currently, the World Health Organization (WHO) considers that it is a global health problem, which is why it is part of the action plan for the prevention and control of noncommunicable diseases.6
Factors Involved in psoriasis.
| Associated factor | Pathophysiological role |
|---|---|
| Drugs (beta-blockers, lithium, antimalarials, nonsteroidal anti-inflammatory drugs, ACE inhibitors, interferons, terbinafine) | Interferes with the immune system, signaling pathways and secretion of cytokines |
| Ultraviolet radiation (e.g., phototherapy) | Immunoregulatory and proapoptotic effect |
| Downregulation of IL-23/Th17 | |
| Cigarette smoking | Oxidative stress |
| Diet, obesity | Overexpression of cytokines |
| Infections (beta hemolytic streptococcus, Staphylococcus aureus, Porphyromonas gingivalis, Candida albicans, Chlamydia psittaci, hepatitis C virus and HIV | Induction of autoimmunity, breaking of immune tolerance |
| Genetics | Predisposition to psoriasis |
| PSORS and HLACw6, among others | |
| Stress | Alteration in the regulation of the immune system and abnormal activation of T cells |
ACE inhibitors: angiotensin-converting enzyme inhibitors; HIV: (human immunodeficiency virus).
Beyond the skin and nail manifestations, it is a systemic disease that is associated with multiple comorbidities.7 Patients with Pso have about 2-fold increased risk of presenting type 2 diabetes mellitus8 and other metabolic disorders related to insulin resistance (obesity, dyslipidemia, metabolic syndrome), and a higher risk has been found in those patients with greater extent and severity of cutaneous involvement.9 Likewise, there is an increase in the risk of high blood pressure and cardiovascular disease, inflammatory bowel disease, depression and cancer (mainly skin tumors, lymphoma) among others.10–12
Among the etiopathogenic mechanisms, genetic predisposition and interaction with the environment have been proposed as disease-initiating events. There are genetically programmed interactions, mainly those genes involved in the skin barrier function (LCE3, DEFB4, GJB2), in addition to genes that participate in the innate immune response such as the nuclear factor-κB signaling cascade (TNFAIP3, TNIP1, NFKBIA, REL, FBXL19, TYK2, NOS2, CARD14) and the signaling of the interleukin 23 (IL-23)/IL-17 pathway (HLA-C, IL12B, IL23R, IL23A, TRAF3IP2, ERAP1), as well as other signaling molecules triggered by environmental factors (IFN-α, TNF-α, IL-36, NF).13–15
It has been stated that the established chronic systemic inflammatory response in Pso is similar to that observed in patients with rheumatoid arthritis or Crohn’s disease,16 due to the persistent elevation of non-specific serum inflammatory biomarkers such as C-reactive protein and haptoglobin.17,18 In addition, an increase in serum levels of TNF-α, IFN-γ, IL-6, IL-8, IL-12, IL-1819 and IL-2620 is evidenced. This systemic inflammation leads to the process of endothelial dysfunction and accelerated atherosclerosis, through the development of atheromatous plaques in the intimal layer of the arteries, an underlying process that ultimately triggers the development of cardiovascular events. This atherogenesis is caused by chronic low-grade inflammation as a result of an interaction between immune mechanisms and metabolic abnormalities within the vessel wall.21
Through this concept of systemic inflammation-accelerated atherosclerosis, the hypothesis of psoriatic march has been reached, a term coined by Boehncke et al. in 2010,22 which summarizes the intricate relationship between Pso, chronic inflammation and cardiovascular risk, as well as the bidirectional pathway between Pso and systemic inflammation, with the subsequent induction of insulin resistance, endothelial dysfunction, atherosclerosis and cardiovascular event (stroke and myocardial infarction13,14,22,23) (Fig. 1).

Psoriatic march, sequence of pathophysiological events: 1st step: genetic predisposition; 2nd step: interaction with the environment and induction of innate and adaptive immune response; 3rd step: expression of the disease; 4th step: chronic systemic inflammation that leads to insulin resistance, endothelial dysfunction, accelerated atherosclerosis and increased cardiovascular risk.
From the beginning, Pso was understood as a hyperkeratotic cutaneous disease, however, this concept has changed over time, and today it is considered a systemic disease mediated by T cells.23 It has been found that there is an alteration in the Th1/Th2 ratio, with a predominance of the Th1, Th17 y Th22 cellular immune response, favored by a marked increase in the production of IL-23 generated by keratinocytes, antigen-presenting cells, Langerhans and intestinal cells, which polarizes the response towards Th17 (responsible for the production of IL-17 A, mainly),11,24 which stimulates the secretion of IL-1, IL-6, TNF-α, and gives rise to the inflammatory cascade that will produce the activation and proliferation of keratinocytes, the release of chemokines that promote the recruitment of inflammatory cells in the lesion, and that favor angiogenesis and hypervascularization in the papillary dermis.25
Like in patients with obesity and coronary heart disease, elevated serum levels of TNF-α, IL-6, adhesion molecules such as the cell adhesion molecule 1 (ICAM-1) and E-selectin, angiogenic factors such as the endothelial growth factor a (VEG-F) are found, and during the periods of disease activity their local production has been demonstrated in the skin, adipose tissue and cardiovascular tissue.26 These molecules favor the amplification of the inflammatory response, thus facilitating the activation of inflammatory cells (neutrophils, macrophages), as well as the differentiation and survival of cytotoxic T lymphocytes and natural killer cells. Furthermore, they promote angiogenesis and chemotaxis of other inflammatory cells. On the other hand, there is an alteration of the insulin signaling pathway (inhibition of the insulin receptor tyrosine-kinase, activation of the peroxisome proliferator-activated receptor delta, activation of tyrosine phosphatase, activation of adenosine monophosphate protein kinase) and of the lipid metabolism-adipogenesis, that leads to an increase in the adipose tissue, dyslipidemia and epidermal proliferation. With respect to VEG-F, it has been demonstrated that its elevation is persistent over time even in patients without data of clinical activity, and it has been concluded that in Pso there is a chronic low-grade inflammation rather than a recurrent inflammation.27
Another important role in the pathogenesis of the disease is mediated by the memory CD8+ cytotoxic T lymphocytes at the epidermal junction that express the very late antigen 1 (VLA-1), thus binding to type IV collagen, which allows the entry into the epidermis, and contributes to the inflammation through the local release of pro-inflammatory cytokines of the Th1 and Th17 phenotypes.24
Likewise, the proinflammatory role of the activation of the NLRP3 inflammasome, a molecular complex that activates caspase 1 and promotes inflammation through the release of multiple cytokines, has been highlighted. Up-regulation of this complex has been found in Pso, so that caspase-1 is activated and with this, the levels of IL-1β increase, triggering inflammatory responses of the vascular wall that lead to the progression of the atherosclerosis. This complex is activated by various danger signals, such as cholesterol crystals, calcium phosphate crystals and low-density lipoproteins oxidized in macrophages, to initiate inflammatory responses in the atherosclerotic lesion.28,29
Role of the dendritic cellAs is well known, different populations of dendritic cells (DC) are found in inactive human skin and had been divided into 3 subsets: epidermal Langerhans cells (LC), dermal myeloid cells, and plasmacytoid cells.30
DCs are part of the arsenal of antigen-presenting cells, and are responsible for initiating and stimulating both the innate and adaptive immune response. Likewise, they play a crucial role in the early stages of the disease, and during exacerbations, by being activated in the dermis and the epidermis. After this activation, they favor the production of tumor necrosis factor-alpha (TNF-α) and IL-23, which in turn promotes the development of subpopulations of T lymphocytes (polarizing the response towards Th1 and Th17), that will favor vascular and epidermal changes present in Pso, as well as the recruitment of inflammatory cells to the dermis and the epidermis, so that the systemic inflammatory phenomenon is perpetuated.31
In addition, in psoriatic disease, the accumulation up to 30 times higher of myeloid dermal DCs has been described in chronically inflamed tissues compared to normal skin, with an increase in the ratio between DC and T-cells that demonstrates an immature phenotype (CD11c+ BDCA-1−); this favors the proliferation and survival of T cells and the polarization to Th1 and Th17.32 On the other hand, it has been found an alteration in the migration of LCs, which leads to their retention at the dermal level and facilitates a greater inflammatory response.33
Toll-like receptorsToll-like receptors (TLR) are part of type I transmembrane proteins, whose function is the recognition of molecular patterns (PRR), among them proteins, nucleic acids and lipopolysaccharides that play a fundamental role in the cutaneous barrier as part of the innate immune response, either against microorganisms, or participating in autoimmune or inflammatory diseases (such as psoriatic disease).34
TLR2 and TLR3 are constitutively expressed in normal keratinocytes and in greater amounts than other receptors of the same family. The activation of TLR2 leads to the production of TNF-α and IL-6 and also improves the function of the tight junction barrier in the epidermis after the invasion of pathogens. Meanwhile, TLR3 is a fundamental part of the normal repair of the cutaneous barrier after tissue damage, thus inducing the expression and function of tight junction components, and promoting the re-epithelialization, granulation and neovascularization necessary for wound healing.35
Overexpression of TLR2, TLR5 and TLR9 has been demonstrated in Pso. In Asian population it has been found that the single nucleotide polymorphism rs3804099 of TLR2 confers susceptibility to the development of Pso vulgaris.36
TLR2 favors the expression of TLR4 in epidermal and dermal DCs. This TLR2/4 pathway facilitates the nuclear translocation of the nuclear factor kappa B (NF-kB) and the increase in the expression of TNF-α and IL-8 in psoriatic keratinocytes, promoting the inflammatory phenomenon in the plaque, and the latter suggests a determining role of TLR2 in the pathogenesis of Pso.37
Endothelial dysfunctionEndothelial dysfunction consists in an imbalance in the release of vasodilator (nitric oxide, prostacyclin) and vasoconstrictor (endothelin and angiotensin II) factors. In patients with Pso, there has been evidence of an increase in endothelin, apparently produced by the keratinocytes, which alters the balance and favors vasoconstriction and the atherogenic pathway.22
In addition, endothelial dysfunction in Pso has been correlated with obesity. This close relationship has been explained by the adipokines, proteins produced in the adipose tissue whose levels are directly proportional to the size of the adipose cells and the accumulation of adipose tissue. They can have proinflammatory (leptin, resistin, osteopontin, chemerin, WNT5A, among others) or anti-inflammatory (adiponectin, SRFP5, omentin, ghrelin and lipocalin 2) phenotype, and it has been found that patients with Pso have low levels of adiponectin, which constitutes a risk factor for endothelial dysfunction.33
Another important factor in endothelial dysfunction is the increased platelet activation and its association with cardiovascular events, in relation to the interaction with chemokines, mainly chemokine ligands 2 and 5 (CCL2, CCL5),38,39 for this reason, it has been studied as a possible therapeutic target. Thus, in a clinical trial, Garshick et al. showed that low doses of aspirin reduced the production of thromboxane B2 and this reduced the expression of proinflammatory transcription, suggesting a possible beneficial effect in patients with Pso. However, clinical studies with cardiovascular outcomes are required to demonstrate the therapeutic effect.40
Alba et al., in a study on intradermal microdialysis, demonstrated a disturbance in the endothelial function of microcirculation, specifically an alteration in nitric oxide-dependent vasodilation, independent of increased oxidative stress, with a decrease in nitric oxide bioavailability.41 Likewise, it has been found a relationship between IL-23 and endothelial dysfunction. In this sense, Sherlock et al. demonstrated in a murine model that it induces inflammation in the aortic root through the activation of CD3+ CD4− CD8 lymphocytes.42
Obesity and insulin resistanceObesity constitutes a predisposing factor for the appearance of Pso, with an estimated prevalence of 23.5%. It is not only that obesity can precede the Pso, there is a reciprocal relationship between both conditions: when comparing with cohorts of patients without Pso, an increased relative risk for the de novo diagnosis of obesity has been observed in those patients who have Pso (HR: 1.18; 95% CI: 1.14–1.23).43 Furthermore, it constitutes a prognostic factor. Worsening of Pso has been demonstrated in obese patients,15,23 and is associated with an increased risk of moderate to severe cutaneous involvement, measured with PASI (OR: 2.23; 95% CI: 1.63–3.05).44
Sustained systemic inflammation induces insulin resistance. Okin et al. found that inflammation suppresses the enzyme of bile acid biosynthesis CYP7A1, leading to the accumulation of 2 metabolites: farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) of the hepatic mevalonate pathway, and the latter leads to elevated fasting glucose, which is reversible by suppressing FPP and GFPP in mouse models.45 The pro-inflammatory status is deleterious to pancreatic cells. In this sense, Grieco et al. demonstrated that IL-17 produces a proapoptotic state in the pancreatic islets and generate a microenvironment that inhibits the secretion of insulin, thus favoring the hyperglycemic state and the subsequent insulin resistance.46 Boehncke et al. found a directly proportional relationship between the severity of Pso (measured by the PASI) and insulin resistance, with the consequent increased risk of developing type 2 diabetes mellitus.47 Furthermore, in a meta-analysis carried out by Coto-Segura et al. it was found that patients with Pso have a higher risk of developing type 2 diabetes mellitus during follow-up (OR: 1.76; 95% CI: 1.59–1.96).48 In addition to the foregoing, they experience an increased risk of micro- and macrovascular complications associate with the disease.49
It has been found that insulin resistance contributes directly to the epidermal phenotype, causing hyperproliferation and altered differentiation of the keratinocytes. However, the relationship between psoriasis and metabolic diseases has also been evidenced by the known chronic inflammation, and in this sense, it is observed how insulin resistance and factors secreted by adipocytes (adipokines) are driving factors.50 For example, resistance to insulin and to dysfunctional HDL occurs concomitantly with the development of adipose tissue inflammation.51
In addition, Pso has been related to several metabolic risk factors, such as dyslipidemia, non-alcoholic fatty liver and hypertension and cardiovascular events.52,53 Mutairi et al. have demonstrated that patients with moderate-to-severe psoriasis have a higher risk of coronary heart disease (OR: 2.97; 95% CI: 1.88–4.69), obesity (OR: 2.356; 95% CI: 1.934–2.87), type 2 diabetes mellitus (OR: 3.137; 95% CI: 2.675–3.68), arterial hypertension (OR: 3.597; 95% CI: 3.015–4.29), dyslipidemia (OR: 3.379; 95% CI: 2.631–4.34) and metabolic syndrome (OR: 2.619; 95% CI: 2.092–3.279). This risk is directly related to the severity of the skin involvement.54
Adipokines in PsoPro-inflammatory adipokines play a critical role in chronic inflammatory phenomenon, endothelial dysfunction, and accelerated atherosclerosis.55 Leptin, a protein encoded by the obesity gene on chromosome 7, participates in the control of body weight by regulating food intake and caloric expenditure. In addition, it regulates the processes of vasoconstriction mediated by angiotensin II and vasodilation mediated by nitric oxide56; likewise, it regulates the expression of adhesion molecules, angiogenesis and keratinocyte proliferation.57 Obesity induces a state of hyperleptinemia, the cells of the immune system (DC, macrophages, neutrophils, effector and regulatory T cells) have receptors for leptin. Downstream, this stimulation promotes cell proliferation and survival and increases the production of TNF-α, IL-1β, IL-12, IL-17, which favors the proinflammatory phenotype.58
Elevation of leptin has been demonstrated in patients with Pso and Psa compared to healthy individuals and correlates with the disease activity.59 However, the initiation of systemic therapy in Pso (both synthetic and biological) has not demonstrated a decrease in the levels of leptin and other adipokines,60, and this suggests that the chronic inflammatory phenomenon in obesity is a factor independent of Pso. Nevertheless, it is still a topic of intense debate.61
Accelerated atherosclerosisChronic low-grade systemic inflammation, resulting from the interaction between immune mechanism and metabolic abnormalities inside the vessel wall, causes the process of accelerated atherogenesis. A qualitative change in the intimal layer of the arteries (endothelial dysfunction) has been found early in patients with Pso.18
Likewise, inflammation of the aortic root compared with people without Pso has been demonstrated as a surrogate marker of major cardiovascular events.62 This inflammation of the aortic root is directly related to the severity of the stenosis of the epicardial coronary arteries, which confirms the increased risk of cardiovascular events in this population.63
Intimal thickeningThe use of ultrasound for the evaluation of the carotid intima-media thickness provides a useful method for the evaluation of subclinical aterosclerosis.64 In analytical studies, it has been found a relationship between Pso and increased carotid intima-media thickness that contributes to atherosclerosis and subsequent cardiovascular outcomes, as demonstrated by Argote et al., who found and average carotid intima thickening of 0.7 mm measured by ultrasound in 40 patients con Pso.65 In a study of 50 patients with plaque Pso, Abrahão-Machado et al. found a strong positive correlation between the Framingham risk score and the quantitative measurement of the carotid intima-media thickness. This makes it a useful tool for the assessment of cardiovascular risk in patients with Pso.64
Cardiovascular eventsThe association between Pso and increased cardiovascular risk is well known. In 1974 McDonald and Calabresi described the increased incidence of occlusive vascular disease in 253 patients with Pso without specific systemic treatment, and they found that 11.5% of the individuals presented one or more arterial occlusive events (stroke, myocardial infarction, thrombophlebitis and pulmonary embolism),66 and this has been corroborated by several epidemiological studies over time.67–70
It is important to highlight that an increased cardiovascular risk is not only due to intrinsic factors. The diet rich in saturated fatty acids also has a deleterious effect in the amplification of the inflammation in Pso, since it can change the composition of the cell membrane and alter the internal cell signaling, leading to an activation of protein kinase C and MAP kinases. The latter produces an increase in the secretion of IL-6 and IL-8,71,72 which highlights the importance of the diet in the control of the disease. Herbert et al. demonstrated that a diet low in fatty acids and rich in omega 3, together with conventional treatment, reduces the severity of psoriasis.73
An unhealthy lifestyle, coupled with chronic systemic inflammation and increased systemic and local (skin and joint) levels of TNF-α and IL-6,74 is deleterious and favors early atherosclerosis, promoting the occurrence of cardiovascular adverse events.75 Gelfand et al. demonstrated that patients with severe Pso have a 50% increased risk of death, which is maintained even after adjusting for risk factors for mortality (cigarette smoking, body mass index, myocardial infarction, congestive heart failure, peripheral vascular disease, cerebrovascular disease, dementia, chronic lung disease, rheumatological disease, peptic ulcer disease, liver disease, diabetes mellitus and chronic complications, hemiplegia or paraplegia, kidney disease, malignant neoplasm, metastatic solid tumor, and AIDS), being premature between 3 and 6 years, compared with people without Pso.76 Mortality due to cardio-cerebrovascular events is associated with Pso as an independent risk factor.77–79 Raaby et al. demonstrated that the risk depends on the severity and extent of the cutaneous domain: the greater the extension, the higher the risk of cerebrovascular events (HR: 1.10 and 1.38 for moderate and severe psoriasis, respectively; acute myocardial infarction; HR: 1.20 and 1.70 for moderate and severe psoriasis, respectively).80 The foregoing highlights the importance of promptly identifying the patients with extensive involvement with a higher cardiovascular risk who would benefit from early initiation of systemic therapy.
Psoriatic arthritisPsa affects up to 30% of patients with Pso and causes severe physical limitations and disability. Some fundamental factors involved in the development of Psa have been described,80–82 (Table 2Table 2), and like Pso, it entails an increased risk of cardiovascular disease (prevalence of obesity, hypertension, dyslipidemia and diabetes). Polachek et al. demonstrated that patients with Psa experience an increased risk of cardiovascular events (OR: 1.43; 95% CI: 1.24–1.66), acute myocardial infarction (OR: 1.68; 95% CI: 1.31–2.15), stroke (OR: 1.22; 95% CI: 1.05–1.41) and heart failure (OR: 1.32; 95% CI: 1.11–1.57), compared to the general population and comparable to that of Pso with severe involvement. This suggests that, the greater systemic inflammation indicated by the activity of the disease, the higher the cardiovascular risk.83 Among the clinical predictors associated with increased cardiovascular risk are dactylitis (relative risk [RR]: 1.20; 95% CI: 1.10–1.35) and the number of painful joints (RR: 1.94; 95% CI: 1.15–3.27).84
Factors involved in psoriatic arthritis.
| Pathophysiological role | |
|---|---|
| Cigarette smoking | Increases activity and severity of psoriatic arthritis |
| Genetic predisposition | Susceptibility to the development of psoriatic arthritis (HLA-Cw*0602, HLA-B27, HLA-B38, HLA-B39, gene IL23R) |
| Severity of psoriasis | Mediated by Th1 (chronic secretion of TNF-αby macrophages), Th17 and autoreactive cytotoxic CD8+ T lymphocytes (inflammation and tissue damage) |
| β-hemolytic streptococcal infection | Cross-reactivity between these Ag and those present in the skin |
| Obesity | Chronic inflammation, hyperleptinemia (proinflammatory factor) and TNF-α (activation and recruiting of immune cells in the synovium) |
TNF-α: tumor necrosis factor-alpha.
Studies in patients with Psa demonstrate greater endothelial dysfunction,85 subclinical atherosclerosis and arterial stiffness,86 for which the pathophysiological mechanism is similar, at least in part, to what has been exposed in relation to the concept of psoriatic march.
Evaluation and reduction of cardiovascular riskDespite the demonstrated relationship between Pso, Psa and cardiovascular mortality, there is no recommendation aimed at this population to prevent and manage cardiovascular risk, so management is carried out according to the risk management guidelines for the general population, which can potentially underestimate the risk and limit aggressive interventions in this population. However, understanding psoriasis, its complex pathophysiology and its relationship with multimorbidity allows us to have more ambitious objectives to achieve better control of the activity in the cutaneous, articular (both peripheral and axial), enthesitis and dactylitis domains, intensive and early treatment of comorbidities, and thus impact cardiovascular outcomes.87
In relation to the use of systemic therapy and its impact on cardiovascular outcomes in Pso, it is still a matter of study, unlike other systemic inflammatory diseases such as rheumatoid arthritis, for which the impact of the use of synthetic and biological therapy on the reduction of cardiovascular risk has been demonstrated.88,89 In Pso, a meta-analysis found limited evidence regarding the reduction in the risk of all cardiovascular events with the use of systemic therapy (non-steroidal anti-inflammatory drugs, corticosteroids, methotrexate, anti-TNF-α),83 while in Psa, Roubille et al. demonstrated that the use of systemic therapy (biological and synthetic) reduced the risk of all cardiovascular events (HR: 0.75; 95% CI: 0.63–0.91). From the clinical point of view, it is easy to intuit that the adequate control of the activity in the domains of the disease, along with the control of traditional modifiable risk factors, has a significant impact on the reduction of cardiovascular risk that will be demonstrated in the long term.89
In relation to the use of statins, the mainstay pharmacological group for the treatment of hyperlipidemia, an immunomodulatory effect has been demonstrated (they favor the Th1-mediated response, inhibit the induction of the major histocompatibility complex II, prevent the release of cytokines and the degranulation of mast cells, and inhibit the interactions between proinflammatory chemokines). For this reason, its usefulness in Pso has been studied. A meta-analysis that included 5 studies demonstrated in patients with severe Pso who received statins vs. placebo, a statistically significant improvement in the PASI, a difference in means of 2.76, (95% CI: 0.49–5.04). However, it is not clear whether the routine use of statins in patients with Pso without hyperlipidemia has an impact on the reduction of the risk of coronary heart disease or other atherosclerotic diseases. Theoretically, they should benefit them since it is proven that they reduce the risk of clinical manifestations of the atherosclerotic process in patients with high cardiovascular risk; however, well-designed studies that demonstrate the long-term clinical impact with daily use are required.90
Regarding the use of acetylsalicylic acid, additional research is needed to evaluate its effectiveness in the primary prevention and reduction of cardiovascular risk in psoriasis.28
In addition to the therapeutic measures previously described, adequate control of traditional risk factors is recommended, as well as weight control, smoking cessation and physical exercise, which has been demonstrated to reduce the concentration of cytokines such as TNF-α, IL6, IL8 and monocyte chemoattractant protein, as well as CRP, which may lead to a decrease in cardiovascular risk and in the severity of the psoriasis, along with an improvement of the quality of life.91–93
ConclusionsPsoriasis is a complex disease. Beyond the skin involvement, it should be understood as a systemic disease that is associated with a higher risk of cardiovascular mortality. The term psoriatic march makes reference to the relationship between Pso, disease activity, systemic inflammation, insulin resistance, accelerated atherosclerosis and cardiovascular risk. The pathophysiological mechanisms that explain these phenomena are variable and have not been fully elucidated. Understanding these mechanisms of inflammation is of vital importance to influence the systemic and multimodal management of the disease. Individual assessment of cardiovascular risk in Pso and Psa, as well as intensive treatment of the disease activity and traditional cardiovascular risk factors, are imperative to reduce morbidity and mortality, however, more evidence on the assessment of risk and the impact of these interventions on the cardiovascular outcomes is required.
FundingThe authors declare that they have not received funding for this work.
Conflict of interestThe authors declare that they have no conflict of interest.
