




El síndrome de activación macrofágica (SAM) es una grave complicación de varias entidades reumáticas entre las que se encuentran la artritis idiopática juvenil sistémica, enfermedad de Still y lupus eritematoso sistémico. Este síndrome forma parte de las linfohistiocitosis hemofagocíticas adquiridas y constituye una enfermedad potencialmente mortal, con dificultad en su identificación y carencia de consensos en cuanto a su manejo. Describimos una serie de casos de pacientes con SAM, exponiendo su proceso diagnóstico, su relación con las enfermedades reumáticas de base, su seguimiento y tratamiento, así como los resultados de diferentes esquemas de manejo.
Macrophage activation syndrome (MAS) is a serious complication of several rheumatic disorders, among which are the systemic juvenile idiopathic arthritis, Still's disease and systemic lupus erythematosus. This syndrome is part of the Acquired Haemophagocytic Lymphohistiocytoses, and is a potentially fatal disease, with difficulty in its identification and a lack of consensus regarding its management. A series of cases are describe of patients with macrophage activation syndrome, explaining their diagnostic process, their relationship with rheumatic diseases, their monitoring, and treatment, as well as the results of different management schemes.
El síndrome de activación macrofágica (SAM) es una condición potencialmente fatal que se encuentra en el grupo de las linfohistiocitosis hemofagocíticas adquiridas. El término SAM se utiliza para referirse al síndrome asociado a entidades reumatológicas, principalmente la artritis idiopática juvenil sistémica, la enfermedad de Still y el lupus eritematoso sistémico (LES)1,2. El SAM se caracteriza por tratarse de un proceso inflamatorio agudo, que se presenta con fiebre alta de difícil remisión, hepatoesplenomegalia, linfadenopatías, manifestaciones hemorrágicas y disfunción del sistema nervioso central, además de la alteración de varios exámenes de laboratorio incluyendo pancitopenia, hipofibrinogenemia, hipertrigliceridemia, disminución de la VSG e hiperferritinemia, así como la observación de macrófagos fagocitando células hematopoyéticas en el aspirado y biopsia de médula ósea (AMO)3,4. Este síndrome se puede presentar a cualquier edad, pero es más común en la quinta década de la vida y en mujeres5,6. Es producido por una respuesta inmunitaria anormal de origen multifactorial, con un aumento de la activación de linfocitos T y macrófagos. La herencia de genes defectuosos involucrados en el control de la citólisis conduce a una actividad citolítica de células NK; estas células no logran eliminar las células tumorales o infectadas, por lo que la proliferación de la población de células T citotóxicas induce la activación y la proliferación de macrófagos tisulares (histiocitos), produciendo una tormenta de citocinas, llevando a un estado proinflamatorio con la producción aumentada de interferón gamma, factor de necrosis tumoral alfa, IL-1, IL-6 y una disminución de IL-107.
El SAM suele presentar factores precipitantes para su aparición, entre los que podemos encontrar exacerbaciones de la enfermedad reumatológica de base, uso de medicamentos como antiinflamatorios no esteroideos, infecciones bacterianas, micóticas o virales, siendo la más importante la infección por virus de Epstein-Barr (EBV), malignidad y trasplantes8. Describimos una serie de casos de pacientes con SAM, exponiendo su proceso diagnóstico, su relación con las enfermedades reumáticas de base, su seguimiento y tratamiento, así como los resultados de diferentes esquemas de manejo.
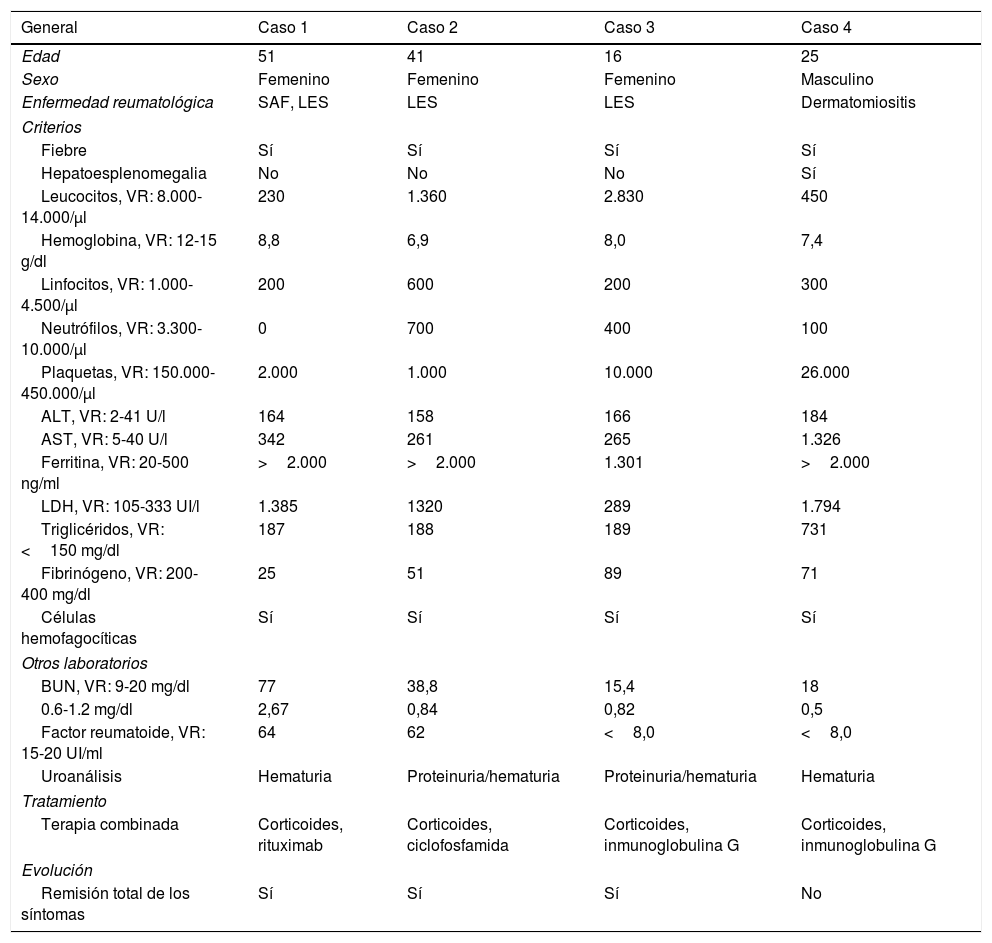
Presentación de los casosCaso 1. Paciente femenina de 51 años, mestiza, con diagnóstico de síndrome de anticuerpos antifosfolípidos cumpliendo criterios clínicos: morbilidad gestacional (óbito fetal> 10 semanas) y tromboembolismo pulmonar submasivo (gammagrafía V/Q) y criterios de laboratorio (anticardiolipina IgM: 154 positiva, β-2 glucoproteína 1 IgM: 31,9 positivo, β-2- glucoproteína 1 IgG: 33,9 positivo); secundario a LES con criterios SLICC 2012 sustentado en: aftas orales, leucocitopenia/linfopenia, serositis (derrame pleural bilateral), anticuerpos antinucleares (ANA): 1/640, anti-DNA: 141,96, consumo de complemento, compromiso renal (proteinuria> 500mg/24h) y Coombs directo positivo (sin anemia hemolítica). Ingresa a urgencias en junio del 2017 presentando fiebre, aftas orales, disnea de medianos esfuerzos y fenómeno de Raynaud. En el examen físico se documenta fiebre de 38,7°C, fenómeno de Raynaud y sobrepeso (índice de masa corporal: 28,3). Los estudios de laboratorio reportaban síndrome anémico de tipo macrocítico, leucocitopenia, linfopenia, neutropenia, trombocitopenia, VSG elevada, alteración de función renal, elevación de transaminasas, factor reumatoide positivo, hiperferritinemia, consumo de complemento y Coombs directo positivo (tabla 1). El AMO que describía infiltrado de células monomórficas e inmunohistoquímica con patrón de linfoma folicular y macrófagos fagocitando células hematopoyéticas.
Características clínicas, serológicas y tratamiento de los pacientes
| General | Caso 1 | Caso 2 | Caso 3 | Caso 4 |
|---|---|---|---|---|
| Edad | 51 | 41 | 16 | 25 |
| Sexo | Femenino | Femenino | Femenino | Masculino |
| Enfermedad reumatológica | SAF, LES | LES | LES | Dermatomiositis |
| Criterios | ||||
| Fiebre | Sí | Sí | Sí | Sí |
| Hepatoesplenomegalia | No | No | No | Sí |
| Leucocitos, VR: 8.000-14.000/μl | 230 | 1.360 | 2.830 | 450 |
| Hemoglobina, VR: 12-15 g/dl | 8,8 | 6,9 | 8,0 | 7,4 |
| Linfocitos, VR: 1.000-4.500/μl | 200 | 600 | 200 | 300 |
| Neutrófilos, VR: 3.300-10.000/μl | 0 | 700 | 400 | 100 |
| Plaquetas, VR: 150.000-450.000/μl | 2.000 | 1.000 | 10.000 | 26.000 |
| ALT, VR: 2-41 U/l | 164 | 158 | 166 | 184 |
| AST, VR: 5-40 U/l | 342 | 261 | 265 | 1.326 |
| Ferritina, VR: 20-500 ng/ml | >2.000 | >2.000 | 1.301 | >2.000 |
| LDH, VR: 105-333 UI/l | 1.385 | 1320 | 289 | 1.794 |
| Triglicéridos, VR: <150 mg/dl | 187 | 188 | 189 | 731 |
| Fibrinógeno, VR: 200-400 mg/dl | 25 | 51 | 89 | 71 |
| Células hemofagocíticas | Sí | Sí | Sí | Sí |
| Otros laboratorios | ||||
| BUN, VR: 9-20 mg/dl | 77 | 38,8 | 15,4 | 18 |
| 0.6-1.2 mg/dl | 2,67 | 0,84 | 0,82 | 0,5 |
| Factor reumatoide, VR: 15-20 UI/ml | 64 | 62 | <8,0 | <8,0 |
| Uroanálisis | Hematuria | Proteinuria/hematuria | Proteinuria/hematuria | Hematuria |
| Tratamiento | ||||
| Terapia combinada | Corticoides, rituximab | Corticoides, ciclofosfamida | Corticoides, inmunoglobulina G | Corticoides, inmunoglobulina G |
| Evolución | ||||
| Remisión total de los síntomas | Sí | Sí | Sí | No |
LES: lupus eritematoso sistémico; SAF: síndrome de anticuerpos antifosfolípidos; VR: valor de referencia.
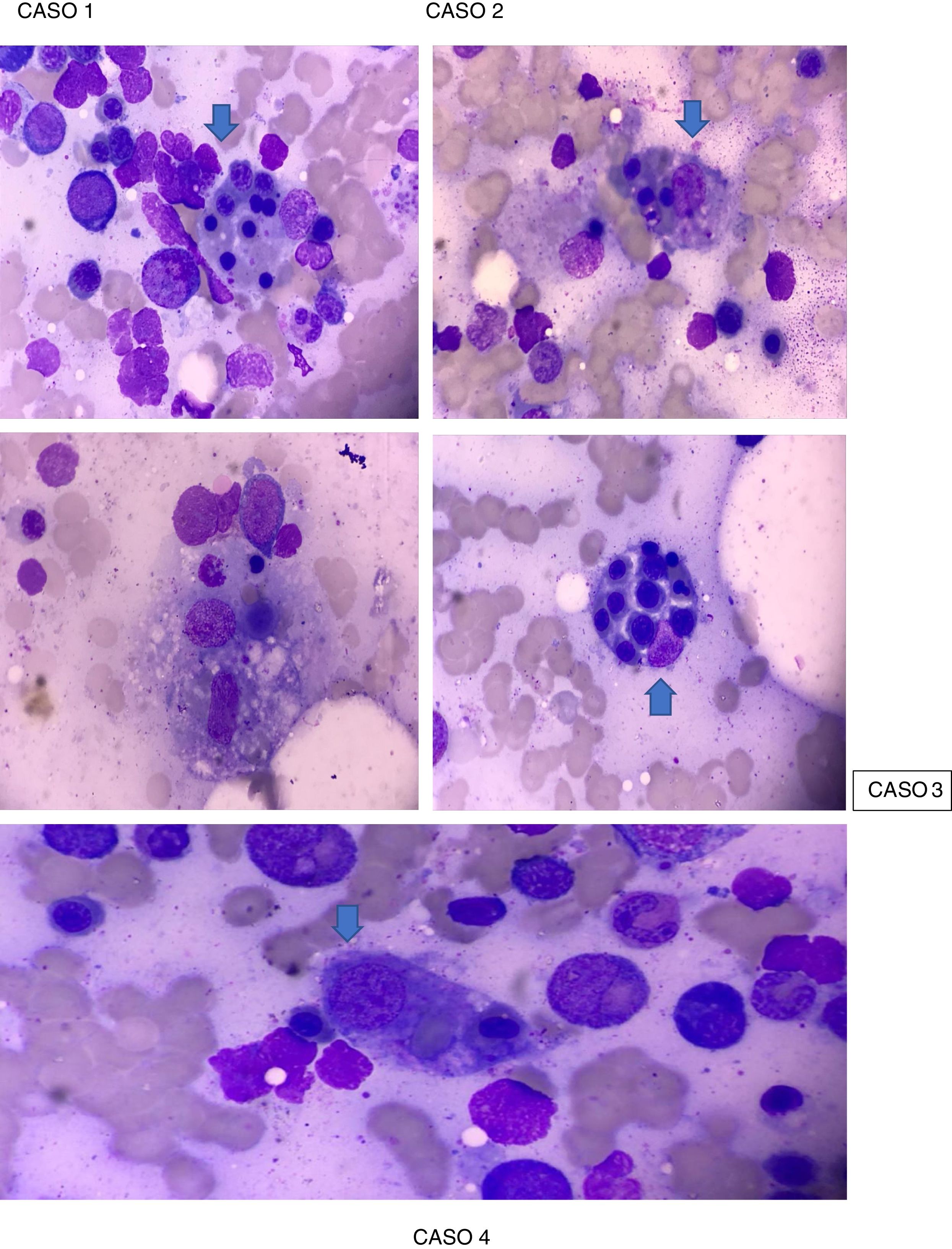
La paciente presentó criterios de Parodi para SAM (tabla 2), sustentado en criterios clínicos: fiebre y criterios de laboratorio: citopenia en 2 o más líneas celulares, hemoglobina (Hb) <9,0g/dl, elevación de transaminasas e hiperferritinemia con un AMO que reveló macrófagos fagocitando células hematopoyéticas (fig. 1). Se ingresó en la Unidad de Cuidados Intensivos (UCI), con aislamiento protector. Se inició manejo farmacológico con pulsos de metilprednisolona 500mg IV durante 3 días consecutivos seguido de prednisona por vía oral a dosis de 1mg/kg/día máx., 60mg/día, hidroxicloroquina 400mg/día, profilaxis para Pneumocystis jirovecii con trimetoprima/sulfametoxazol 960mg 3 veces por semana, presentando mejoría franca, buena modulación de su respuesta inflamatoria, con cultivos negativos, descenso de reactantes de fase aguda y mejoría de parámetros hematológicos (exceptuando plaquetas). Se consideró administrar rituximab, lo cual mostró un ascenso considerable en el recuento de plaquetas> 100.000/ml tras la segunda dosis, sin presentar reacciones infusionales. Dadas la estabilidad hemodinámica y la compensación de su enfermedad clínica, se da de alta por la especialidad de Reumatología con indicación de dosis de rituximab en 6 meses, fondaparinux 7,5mg subcutáneo cada día y control ambulatorio por Reumatología y Hematología.
Criterios diagnósticos de Parodi para síndrome de activación macrofágica
| Criterios clínicos |
| Fiebre>38°C |
| Hepatomegalia (≥ 3cm por debajo del reborde costal) |
| Esplenomegalia (≥ 3cm por debajo del reborde costal) |
| Manifestaciones hemorrágicas |
| Disfunción del sistema nervioso central |
| Criterios de laboratorio |
| Citopenia en 2 o más líneas celulares (hemoglobina <9,0 g/dl, leucocitos: <4.000/μl, plaquetas: <100.000/μl, neutrófilos: <1.000/μl) |
| Aumento aspartato aminotransferasa (> 40 unidades/l) |
| Aumento del lactato deshidrogenasa (> 567 unidades/l) |
| Hipertrigliceridemia> 178 mg/dl |
| Hipofibrinogenemia> 150 mg/dl |
| Ferritina>500 μl/l |
| Criterio histopatológico |
| Evidencia de macrófagos hemofagocíticos en médula ósea |

Caso 2. Paciente femenina de 41 años, mestiza, con diagnóstico de LES presentando criterios SLICC 2012, sustentado en: alopecia, ANA de ½.560 patrón homogéneo, leucocitopenia/linfopenia, trombocitopenia, serositis (derrame pleural), sinovitis clínica en manos y rodillas, Coombs directo positivo, anemia hemolítica, compromiso renal (proteinuria> 500mg/24h), quien ingresa en marzo del 2018 por fiebre de 2 meses de evolución y hemoptisis. En el examen físico presentaba fenómeno de Raynaud bifásico en manos, livedo reticularis en pies bilateral y discreto rash generalizado de predominio en el tórax anterior. Es ingresada para manejo hospitalario y se le realizan exámenes de laboratorio (tabla 1), con lo que se encuentra que cumple criterios de Parodi para SAM (tabla 2), sustentado en criterios clínicos: fiebre y manifestaciones hemorrágicas y criterios de laboratorio: citopenia en 2 o más líneas celulares, Hb <9,0g/dl, aumento de aspartato aminotransferasa>40 e hiperferritinemia, AMO que reveló macrófagos fagocitando células hematopoyéticas (fig. 1). No se evidenció la presencia de foco infeccioso. Se inició esquema de 3 días de metilprednisolona 500mg UCI en combinación con una dosis de ciclofosfamida 500mg IV, mostrando una mejoría clínica sustancial, por lo que se inició prednisona por vía oral a dosis de mantenimiento de 1mg/kg/día, presentando remisión total de los síntomas.
Caso 3. Paciente femenina de 16 años, mestiza, con diagnóstico de LES, presentando criterios SLICC 2012, sustentado en compromiso cutáneo (rash malar), úlceras orales, alopecia no cicatricial, manifestaciones articulares y alteraciones hematológicas (leucocitopenia, linfopenia y trombocitopenia severa), y criterios de laboratorio (ANA 1/5.120 patrón moteado, anti-DNA 1/80, anti-Sm positivo, anticardiolipina IgM positivo y Coombs directo positivo), quien ingresa en octubre del 2018 por cuadro clínico de 3 días de evolución consistente de fiebre, epistaxis y gingivorragia asociado a periodos de desorientación. Es ingresada a hospitalización donde se le realizan exámenes de laboratorio (tabla 1), encontrándose que cumple criterios de Parodi para diagnóstico de SAM (tabla 2), sustentado en: disfunción neurológica, fiebre y manifestaciones hemorrágicas, además de criterios de laboratorio que incluyen: pancitopenia severa, aspartato aminotransferasa severamente elevada, lactato deshidrogenasa (LDH) elevada, hipofibrinogenemia, hipertrigliceridemia e hiperferritinemia y AMO que reveló macrófagos fagocitando células hematopoyéticas (fig. 1). Se inició manejo con pulsos de metilprednisolona 500mg IV, durante 3 días consecutivos, seguido de prednisona por vía oral a dosis de 1mg/kg/día máx. 60mg/día en combinación con IgG a dosis de 2mg/k/día por 4 días consecutivos y estimulador de colonia de granulocitos, presentando una evolución clínica favorable con posterior remisión de los síntomas.
Caso 4. Paciente masculino de 25 años, blanco, conocido por la especialidad de Reumatología por cuadro clínico que inició en junio del 2017 con debilidad muscular generalizada, artralgia de muñecas asociado a edema, limitación para la movilidad, rigidez matutina y posterior dolor en articulaciones MCF, IFP, rodillas, tobillos, hombros, además de fiebre de predominio matutino diario, sin rash, caída del cabello con áreas de alopecia, caída de las uñas y aparición de lesiones en la piel en áreas extensoras de articulaciones MCF, IFP, codos, mentón y superficie malar, descritas como placas eritematodescamativas, fiebre, epistaxis con hallazgos de úlceras nasales y hematoquecia. Durante la hospitalización se descartaron enfermedades como LES (función renal normal, C3 y C4 normales, ANA, ENA), anticuerpos antifosfolípidos negativos) y esclerosis sistémica (anti-SCL-70 negativo y anticuerpos anticentrómeros negativos). Entre las hipótesis diagnósticas se consideró una probable miopatía inflamatoria de tipo dermatomiositis cumpliendo criterios de Bohan y Peter, por hallazgos en la piel, debilidad muscular simétrica generalizada, electromiografía+neuroconducción con patrón miopático y resonancia magnética que reportó miositis de tipo inflamatorio. Sin embargo, las enzimas musculares (CK total, LDH, TGO, TGP, aldolasa) mostraban resultados normales y la biopsia muscular fue negativa. El paciente estaba en tratamiento con prednisolona 20mg y azatioprina 50mg indicada en la consulta externa, en combinación con terapia física. Se le realizó tamización para enfermedad de Pompe siendo este negativo y se le solicitó una segunda biopsia de músculo semitendinoso izquierdo reportando atrofia no específica a establecer por marcadores de inmunoperoxidasa, los cuales no pudieron ser realizados por no autorización de su EPS.
Durante la hospitalización en septiembre del 2019, el paciente se deteriora en su estado clínico presentando taquicardia de 120 lpm y ruidos cardiacos que impresionaba frote pericárdico. Se actualizan los estudios de laboratorio (tabla 1) reportando leucocitopenia, linfopenia, neutropenia, anemia microcítica-normocrómica, trombocitopenia, elevación de transaminasas, nitrógeno ureico en sangre, creatinina, IgG, además de factor reumatoide positivo a título bajo (tabla 1). El ecocardiograma reportó ventrículo izquierdo de tamaño normal, función sistólica global y segmentaria normales, fracción de eyección: 60%, insuficiencia mitral trivial y tricuspídea, válvula aórtica de aspecto y funcionamiento normal, cavidades derechas de tamaño normal, derrame pericárdico moderado a severo, sin repercusión hemodinámica. La ecografía de abdomen total mostró lesión focal hepática que por sus características ecográficas sugería como primera posibilidad hemangioma y la tomografía computarizada de tórax reportó adenopatías mediastinales, hepatoesplenomegalia y severo derrame pericárdico hipodenso. Con estos hallazgos, se consideró que cumplía criterios Parodi para el diagnóstico de SAM (tabla 2), sustentado en: fiebre>38°C, hepatoesplenomegalia, citopenias en> 2 líneas celulares, Hb <9g/dl, aumento de transaminasas, hiperferritinemia, manifestaciones hemorrágicas y confirmación por AMO, que reveló macrófagos fagocitando células hematopoyéticas (fig. 1). En el manejo intrahospitalario se indicó aislamiento protector, amplio esquema antibiótico y antifúngico, pulsos de metilprednisolona 500mg IV durante 3 días consecutivos seguido de prednisona por vía oral a dosis de 1mg/kg/día máx. 60mg/día en combinación con IgG a dosis de 2mg/k/día por 4 días consecutivos, presentando una evolución clínica favorable. Posteriormente, fue valorado por Hematología, que sospecha un síndrome linfoproliferativo asociado, sugiriendo traslado a un centro oncológico, donde a los pocos días fallece.
DiscusiónLas linfohistiocitosis hemofagocíticas engloban un grupo heterogéneo de enfermedades que pueden presentarse a cualquier edad. La forma genética o primaria se manifiesta antes del año de vida e incluye defectos genéticos conocidos, por ejemplo, perforina, y defectos genéticos desconocidos, deficiencias inmunológicas como síndrome de Chediak-Higashi, síndrome de Griscelli, síndrome linfoproliferativo ligado al cromosoma X, y la forma adquirida o secundaria puede verse en cualquier grupo etario asociada con procesos infecciosos, productos endógenos, oncológicos, reumatológicos o inmunológicos9. El SAM forma parte de las linfohistiocitosis hemofagocíticas adquiridas y constituye una enfermedad potencialmente mortal, con dificultad en su identificación y carencia de consensos en cuanto a su manejo.
Describimos una serie de casos de pacientes con diagnóstico de SAM clasificados por los criterios Parodi (tabla 2), los cuales presentan una sensibilidad del 92,1% y especificidad del 90,9%. Para el diagnóstico se requiere la presencia simultánea de al menos un criterio clínico y por lo menos 2 criterios de laboratorio. La biopsia de médula ósea puede ser necesaria solo en casos dudosos, lo cual es una ventaja de estos criterios con respecto a otros criterios clasificatorios propuestos para la linfohistiocitosis hemofagocítica10.
Nuestros pacientes cumplían más de 5 criterios y presentaban, además, diferentes enfermedades reumatológicas de base, entre otras enfermedades asociadas. Es importante recalcar que, aunque el SAM es encontrado principalmente como complicación del LES, la artritis idiopática juvenil y la enfermedad de Still, es también posible encontrarlo en otras enfermedades reumatológicas y que su aparición puede ser desencadenada por múltiples causas, como pueden ser cuadros infecciosos y uso de medicamentos, o por procesos de tipo neoplásico, entre otros. Sin embargo, en muchos casos no se logra identificar con certeza el agente etiológico asociado, debido a que la agudeza del cuadro clínico acapara la atención del clínico, ignorando que el manejo de esos desencadenantes es un componente fundamental de su tratamiento. Egües Dubuc et al. describieron una serie de 13 casos clínicos de SAM secundario a enfermedades autoinmunes, hematológicas, infecciosas y oncológicas, encontrando el síntoma de fiebre como único criterio en común. La esplenomegalia o la hepatomegalia estuvo presente en 12/13 pacientes estudiados11.
El SAM es potencialmente letal y sus máximas manifestaciones pueden aparecer de manera aguda, llevando rápidamente a la aparición de falla multiorgánica que requiere el ingreso en una UCI, reportándose tasas de mortalidad del 8 al 24% en casos asociados a eventos reumáticos o infecciones y siendo mayor al acompañarse de malignidad. Factores agravantes en estos pacientes incluyen la presencia de neutropenia asociada, que es rara en la historia natural del SAM, donde las líneas granulares no suelen estar alteradas, y la aparición de infecciones oportunistas, lo que no solo empeora el pronóstico del paciente, sino que dificulta su manejo. Es imperativo, entonces, recalcar la importancia de considerar este síndrome en pacientes reumatológicos, ya que suele ser una entidad subdiagnosticada, mayormente debido a su desconocimiento y a la falta de marcadores específicos para la enfermedad.
El objetivo terapéutico se basa en detener el proceso inflamatorio. Debido a que no se cuenta con estudios controlados para el tratamiento del SAM, el tratamiento se basa en la experiencia empírica, siendo el eje principal el uso de glucocorticoides parenterales en altas dosis, principalmente una terapia de pulsos de metilprednisolona por 3 días consecutivos seguido de prednisona por vía oral. En casos no controlados con glucocorticoides, se hace asociación con una terapia inmunosupresora como la ciclosporina A (2-7mg/kg/día)12. El uso de ciclofosfamida, inmunoglobulinas (Ig), plasmaféresis y etopósido como terapia inicial ha tenido resultados conflictivos. Sin embargo, cabe mencionar que el uso de Ig IV en nuestros pacientes, en combinación con el uso de corticoides, mostró resultados beneficiosos, presentando una evolución clínica favorable con posterior remisión de los síntomas.
En casos de malignidad, se debe seguir un tratamiento específico para el tipo de neoplasia, usando etopósido como parte del régimen o rituximab en casos de linfoma13. Si es debido a una infección, se debe seguir el tratamiento específico correspondiente. Si el SAM resulta refractario a cualquiera de los esquemas antes mencionados o si el agente causal es el EBV, el protocolo de tratamiento de linfohistiocitosis hemofagocíticas es de utilidad13,14. La anakinra y el infliximab pueden ser beneficiosos en estos casos refractarios y el uso de Ig IV y el rituximab han mostrado buenos resultados en presencia de EBV, citomegalovirus y en pacientes con SAM secundario a LES.
ConclusiónEl SAM es una entidad potencialmente mortal, que puede llevar a falla multiorgánica de manera aguda y que es una complicación de algunas enfermedades reumatológicas, frente a la presencia de desencadenantes, que cursa principalmente con fiebre alta, hepatoesplenomegalia, manifestaciones neurológicas, hemorrágicas y alteraciones de laboratorio. Si bien no presenta parámetros fácilmente identificables para su diagnóstico, debe ser considerada y estudiada en pacientes con factores de riesgo y enfermedad reumatológica con el fin de reducir el riesgo de mortalidad y fomentar su conocimiento y manejo entre los clínicos.
FinanciaciónNinguna.
Conflicto de interesesNo hay conflicto de intereses.
