




Camurati–Engelmann disease is a rare entity due to mutations in the gene encoding the TGF-β. It is characterized by hyperostosis of long bones and skull, accompanied by severe bone pain, and occasionally muscular weakness and a waddling gait. The treatment is based on the use of high doses of glucocorticoids, and in severe cases surgical decompression is indicated. As far as we know, this is the first case reported in Colombia.
La enfermedad de Camurati-Engelmann es una entidad poco común debida a mutaciones en el gen que codifica el TGF-β. Se caracteriza por hiperostosis de huesos largos y cráneo, acompañada de dolor óseo intenso, ocasionalmente debilidad muscular, marcha de pato. El tratamiento se basa en el uso de glucocorticoides en dosis altas y en casos severos la descompresión quirúrgica está indicada. Desde nuestro conocimiento este es el primer caso reportado en Colombia.
Camurati–Engelmann disease (CED), known as progressive diaphyseal dysplasia, is an uncommon disease due to mutations of the transforming growth factor beta (TGF-β); which participates in bone proliferation. Pain in the long bones is the cardinal symptom of CED. There is no curative treatment for the disease, however, a cycle of glucocorticoids in high doses can be given and, occasionally, decompression surgeries of the medullary canal can be performed to help alleviate pain.
We present the case of a 54-year-old woman with CED, with tibial commitment and no other bone manifestations. As far as we know, this is the first case of CED reported in Colombia.
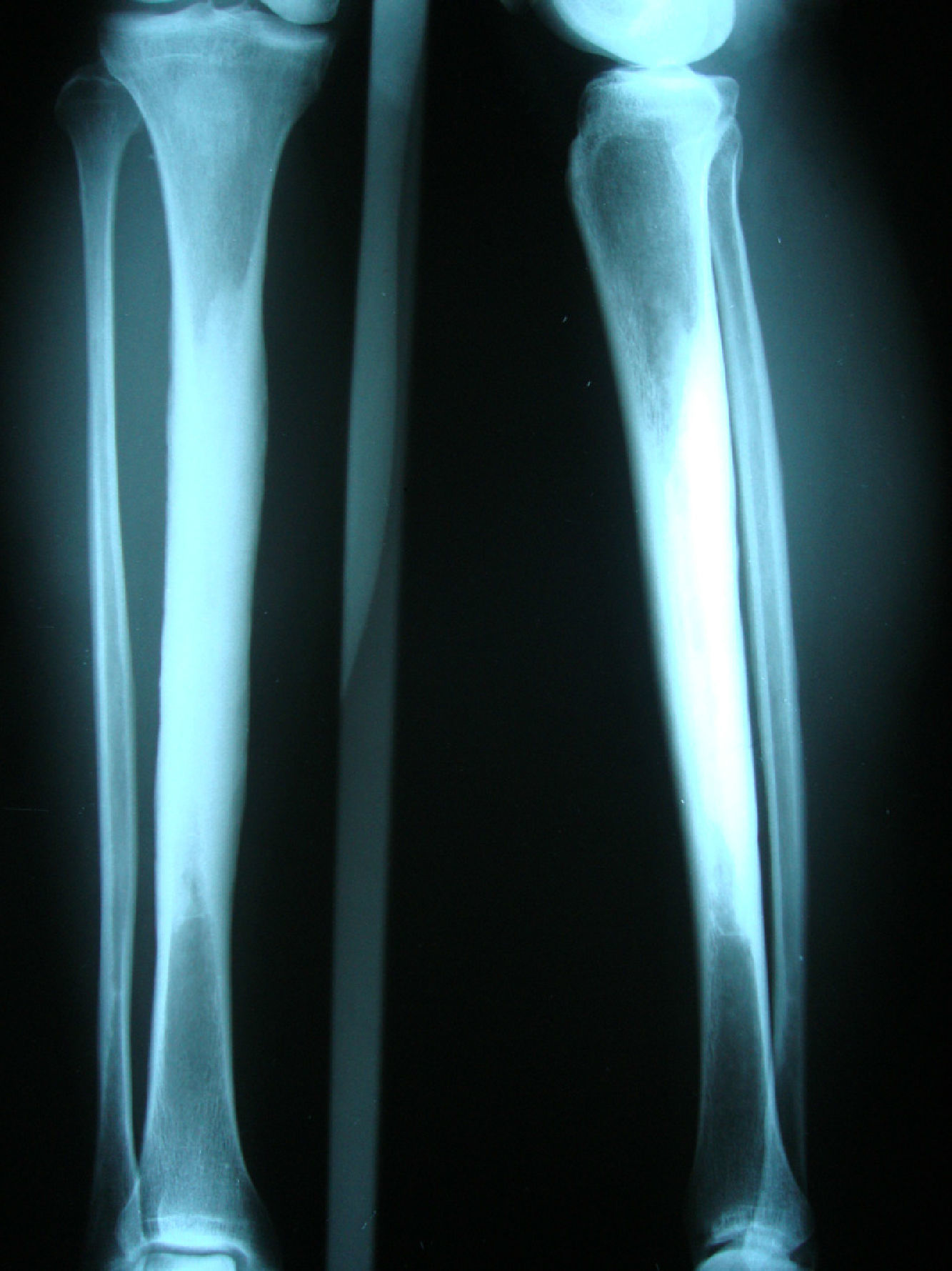
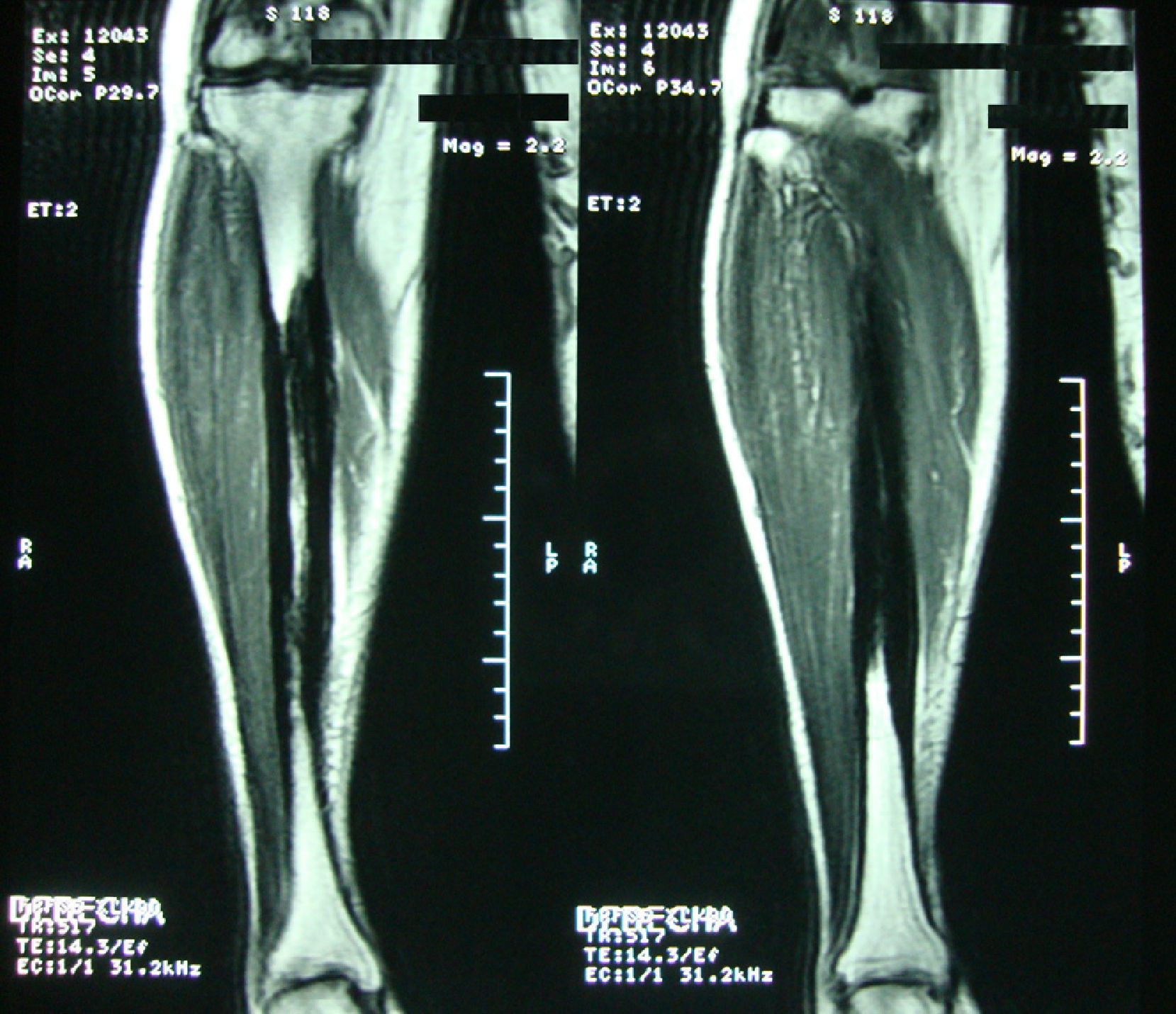
Clinical caseA 54-year-old woman, who 4 years ago had a blunt trauma in the left tibia, since that time she has persistent pain in both lower limbs. In the review of systems she denied any ocular or auditory disorders, previous fractures, neurological deficit or constitutional symptoms. She received medical treatment with NSAIDs without any improvement, and for this reason it was ordered an X-ray of both legs which showed a sclerosing diaphyseal lesion in the middle third with loss of the medullary canal (Fig. 1). The MRI in 2008 confirmed these findings (Fig. 2). The laboratory data such as CBC, ESR, CRP, BUN, creatinine, LDH, ALP, and urinalysis were normal. It was performed a bone biopsy in which it was observed the presence of a large number of small fragments of dense sclerotic bone tissue without any evidence of tumor or inflammatory lesion. The above findings are compatible with the diagnosis of CED. She was assessed by the Service of Orthopedics, who performed a resection of the sclerotic lesions and recanalization of the left tibia.


Over the past 2 years she has had pain in the knees associated with exercise, wrists, elbows, shoulders, and lumbar region without inflammatory signs, insomnia and physical fatigue. The initial physical examination showed a patient with normal height and phenotype, pain to the percussion of the long bones of the lower limbs without bone deformity or neurologic deficit with 15/18 tender points. In addition, it was made a diagnosis of fibromyalgia. The laboratory tests at this time showed normal intact PTH, calcium, serum phosphorus, and proteins with differential, with a C-reactive protein of 96mg/l, and therefore treatment was started with prednisolone 50mg QD, calcium + vitamin D QD, sertraline 50mg QD and trazodone 50mg QD.
The patient was assessed by endocrinology ruling out hyperparathyroidism. In the subsequent controls the glucocorticoids were gradually reduced until being stopped in 6 months, with cessation of the pain to the percussion of the tibia and the fibula, but the patient continued with clear signs and symptoms of fibromyalgia. The acute phase reactants had become normal. It was requested an evaluation by psychiatry for the management of the associated depression. In the subsequent controls the underlying disease has remained stationary.
ReviewThe CED has been known by the names of progressive epiphyseal dysplasia, generalized hyperostosis, congenital multiple hyperostotic disease, sclerosing dysplasia and symmetrical osteosclerosis,1 but the eponym CED is the name most widely known and accepted.
The first description was made by Cockayne in 1920,2 2 years later Camurati suggested its hereditary nature3 and in 1920 Engelmann reported a case characteristic of the disease.4 The CED has an autosomal dominant inheritance with incomplete penetrance and variable expressivity with incomplete penetrance and variable expressivity.5 Recently, the gene responsible for the disease was located in the 19q13.1-13.3 region.6 Mutations in the gene encoding the TGF-β have been found in 90–94% of cases. This gene has 2 exons and the majority of these mutations are in exon 4, followed by exon 1. The 3 most frequent mutant alleles are p Arg210cys, p Arg218His and pCys 225 Arg.7
The transforming growth factor beta (TGF-β) is a bone growth factor, member of the superfamily or the bone morphogenetic proteins;8 it promotes the proliferation, function and survival of the osteoblasts,9 in addition, it has a suppressive action on the differentiation of osteoclasts.10
CED is considered a rare disease with an estimated prevalence of one in a million inhabitants, 200 patients have been identified in the world literature. This pathology has a worldwide distribution based in the report of 24 families from America, Europe, Africa and Oceania. The onset of the disease is in childhood and it usually progresses during the adolescence, but in adulthood its course may be stationary or slowly progressive. The clinical symptoms are present in 74% of patients.11 The limping gait is an early manifestation but it is not the most frequent, occurring in 43%. Progressive muscular atrophy and decreased subcutaneous fat in the limbs also can occur. Easy fatigability is a symptom that has been described in 44%. The cardinal symptom of the disease is bone pain in the extremities in 90% of cases. The patients report that the pain increases with physical activity, stress and cold. Some patients have crises of pain of variable duration (hours-weeks); 58% have pain to bone percussion. Cranial affection occurs in 38% of cases and can be manifested by atrophy of the optic nerve, glaucoma, subluxation of the eyeball, sensorineural or conductive hearing loss in 15%.12 The pathophysiology of this lesions is based on nerve compression or obstruction of the external auditory canal. The sclerosis of the foramen magnum may cause hyperreflexia, weakness, paraparesis and even death.13 The increase in bone mineral density in the diaphyses may increase the risk of fracture with delayed consolidation.14
It has been described a typical disease phenotype characterized by craniomegaly with prominent forehead, exophthalmos, slender limbs with thick and curved bones, marfanoid aspect, flat and valgus feet, lumbar lordosis and scoliosis.
Hepatosplenomegaly, Raynaud's phenomenon, hyperhidrosis of the feet and hands, and delayed dentition and puberty have been reported ocassionally.15 Anemia, leukopenia, increased alkaline phosphatase, parathormone, serum calcium, phosphorus, and erythrocyte sedimentation rate can be observed in the laboratory tests, but is not the norm to find these alterations.14,16–19
The diagnosis of the disease is based on the clinical symptoms and laboratory findings, but is confirmed by the radiological images. The characteristic radiological manifestations are fusiform thickening of the bone cortical. CED has symmetrical affectation and involves both the endosteal and periosteal surfaces. Initially begins in the diaphyses and it extends to the metaphyses, sparing the epiphyses.16,20,21 As a consequence it can be seen a narrowing of the medullary canal with a shape of an Erlenmeyer-flask. The bones affected in order of frequency are: femur, tibia, fibula, humerus, ulna and radius. It can be seen an involvement of the mandible, scapulae, collarbones, pelvis and the skull base. There is rarely commitment of the carpal bones, tarsal bones and phalanges. The characteristic image of the long bones appears in 94% of patients, 63% in the pelvis and 54% in the skull.22
Glucocorticoids are the cornerstone in the treatment of CED, because they increase the apoptosis of the osteoblasts and osteocytes, and also allow the proliferation and differentiation of the osteoclasts.23,24 Conversely, it has been demonstrated that glucocorticoids increase the expression of TGF-β, a deleterious situation in these patients.25 Prednisolone 1mg/kg/day followed by a rapid reduction in the dose in order to avoid side effects is used to control bone pain and fatigue. Losartan is an anti-TGF-β drug and it could be used in patients with arterial hypertension who are intolerant to glucocorticoids.26 Other drugs such as bisphosphonates have been used for the management of CED with disappointing results.27 Surgical treatment s reserved as a decompression mechanism for the external auditory canal. Osteotomies and scrapings of the medullary canal have also been reported with recurrence in some cases.28
In Table 1 are described the main sclerosing bone diseases. In the malignant infantile form of osteopetrosis the bones are dense and fragile. There may be spontaneous bleeding with anemia and recurrent infections. The neurological complications are blindness and deafness. Patients usually die in the first decade of life. The type II form occurs in adolescence and is manifested by fractures, sometimes recurrent, sandwich vertebrae and the bone-within-bone image in the iliac bone. The compression of cranial nerves due to osteosclerosis is rare.29
Main causes of sclerosing bone diseases.
| Osteopetrosis (Albers Schonberg) |
| Osteomesopycnosis (Maroteaux) |
| Osteopoikilosis |
| Osteopathia striata (Voorhoeve's disease) |
| Endosteal hyperostosis(Van Vuchem, Worth syndrome, sclerosteosis) |
| Kenny-Caffey syndrome |
| Metaphyseal dysplasia (Pyle's disease) |
| Progressive diaphyseal dysplasia (Camurati–Engelmann disease) |
| Melorheostosis |
Pycnodysostosis manifests itself in childhood by short stature and a disproportionately large skull. The hands are square with small fingers and pectus excavatum. The recurrent fractures affect, mainly, the lower limbs.30
Osteopoikilosis means spotted bones, which are confused with bone metastases from prostate and breast cancer.31 It is incidentally found by the presence of circular lesions of osteosclerosis. It affects the ends of the short bones, and the metaphyses and epiphyses of the long bones.32 Sometimes is associated with disseminated lenticular dermatofibrosis. The etiology of the disease is associated with mutations in the LEMD3 gene.33
In Van Buchem disease there is an asymmetric enlargement of the jaw during puberty. There are not fractures but it can be pain to the percussion of long bones. Neurological alterations due to compression of cranial nerves are frequent. The disease progresses with age, and therefore, is more common to see radiological findings in elderly patients. In the form of sclerosteosis (Truswell–Hansen disease) the patients are tall and fat with syndactyly and nail dysplasia. The hyperostosis is classically endosteal. In Worth syndrome occur the same findings but it differs from the other 2 variants by the presence of an enlarged and pointed jaw with a widened forehead.34
The presence of linear striations in the long bones is characteristic of the osteopathia striata; and it can be seen cranial sclerosis with nerve palsy.35
In the melorheostosis is seen an eccentric and irregular hyperostosis of the cortex and the medullary canal, like the wax that trickles down the side of a candle.36
In Kenny-Caffey disease there is intramembranous ossification. The main radiological finding is the internal cortical thickening with lack of differentiation of the tables of the skull. The main clinical findings are: short stature, mucosal paleness and microphthalmia.37
Pyle's disease is characterized by widening of the metaphyses of the long bones called Erlenmeyer flask deformity, with growth of the medial parts of the clavicles, the ischium and the pubis.38
Finally, the low back pain that begins in adolescence is the clinical finding of the osteomesopycnosis, due to sclerosis of the terminal plates. It has been described a cystic lesion in the femoral diaphysis. Other long bones or cranial bones have normal characteristics.39
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflict of interestThe authors declare that they have no conflict of interest.
Please cite this article as: Restrepo JP, Molina MP. Enfermedad de Camurati-Engelmann: reporte de un caso y revisión de la literatura. Rev Colomb Reumatol. 2016;23:218–222.
