



La enfermedad de Paget ósea es una enfermedad metabólica del hueso de etiología no esclarecida, que se caracteriza por una fase de resorción aumentada seguida por una fase de osteoformación aberrante. Es frecuente en Europa, Norteamérica, Nueva Zelanda y Australia, pero infrecuente en Asia, Medio Oriente y África. En población colombiana hay reportes de casos. Generalmente cursa asintomática y se diagnostica incidentalmente por hallazgos radiográficos o fosfatasa alcalina elevada. El uso de bifosfonatos favorece el control del recambio óseo y permite prevenir complicaciones como las fracturas. Se presenta una serie de casos en Colombia y una revisión de la literatura.
Paget's disease of the bone is a metabolic bone disease of unknown origin, and is characterised by an increased phase of resorption, followed by an aberrant osteoformation phase. It is common in Europe, North America, New Zealand, and Australia, but infrequent in Asia, the Middle East, Africa, and in the Colombian population there are case reports. It is usually asymptomatic and is diagnosed incidentally by radiographic findings or an elevated alkaline phosphatase. The use of bisphosphonates favours the control of bone turnover and prevents complications such as fractures. A series of cases in Colombia is presented, along with a review of the literature.
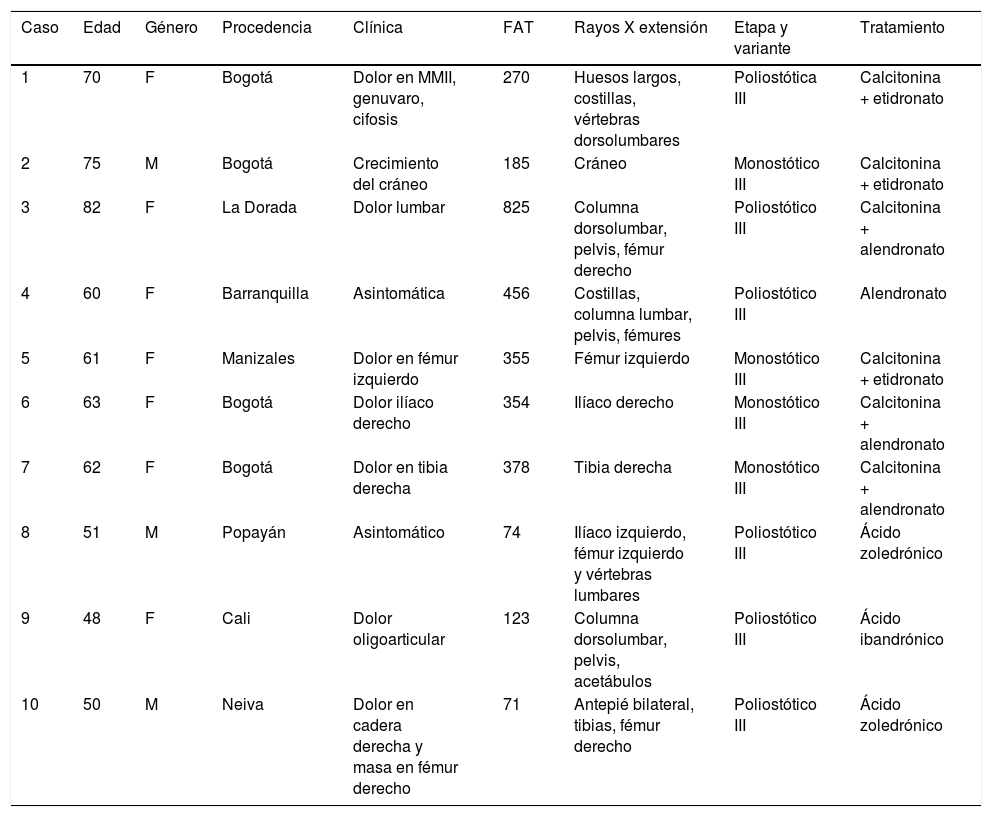
La enfermedad de Paget (EP) es una enfermedad osteometabólica crónica, que afecta uno o varios huesos del cuerpo. De etiología desconocida, con influencia de factores genéticos y ambientales, que desempeñan un papel importante en la fisiopatología de la enfermedad. Su mayor prevalencia está en Europa, concretamente en Gran Bretaña, mientras que en América del Sur hay mucha heterogeneidad y en Colombia es infrecuente1. A continuación se presenta una serie de nuevos casos en Colombia sumados a los previamente publicados en nuestro país2-4 (tabla 1), y se hace una revisión de la literatura.
Casos de EP en Colombia y principales características
| Caso | Edad | Género | Procedencia | Clínica | FAT | Rayos X extensión | Etapa y variante | Tratamiento |
|---|---|---|---|---|---|---|---|---|
| 1 | 70 | F | Bogotá | Dolor en MMII, genuvaro, cifosis | 270 | Huesos largos, costillas, vértebras dorsolumbares | Poliostótica III | Calcitonina + etidronato |
| 2 | 75 | M | Bogotá | Crecimiento del cráneo | 185 | Cráneo | Monostótico III | Calcitonina + etidronato |
| 3 | 82 | F | La Dorada | Dolor lumbar | 825 | Columna dorsolumbar, pelvis, fémur derecho | Poliostótico III | Calcitonina + alendronato |
| 4 | 60 | F | Barranquilla | Asintomática | 456 | Costillas, columna lumbar, pelvis, fémures | Poliostótico III | Alendronato |
| 5 | 61 | F | Manizales | Dolor en fémur izquierdo | 355 | Fémur izquierdo | Monostótico III | Calcitonina + etidronato |
| 6 | 63 | F | Bogotá | Dolor ilíaco derecho | 354 | Ilíaco derecho | Monostótico III | Calcitonina + alendronato |
| 7 | 62 | F | Bogotá | Dolor en tibia derecha | 378 | Tibia derecha | Monostótico III | Calcitonina + alendronato |
| 8 | 51 | M | Popayán | Asintomático | 74 | Ilíaco izquierdo, fémur izquierdo y vértebras lumbares | Poliostótico III | Ácido zoledrónico |
| 9 | 48 | F | Cali | Dolor oligoarticular | 123 | Columna dorsolumbar, pelvis, acetábulos | Poliostótico III | Ácido ibandrónico |
| 10 | 50 | M | Neiva | Dolor en cadera derecha y masa en fémur derecho | 71 | Antepié bilateral, tibias, fémur derecho | Poliostótico III | Ácido zoledrónico |
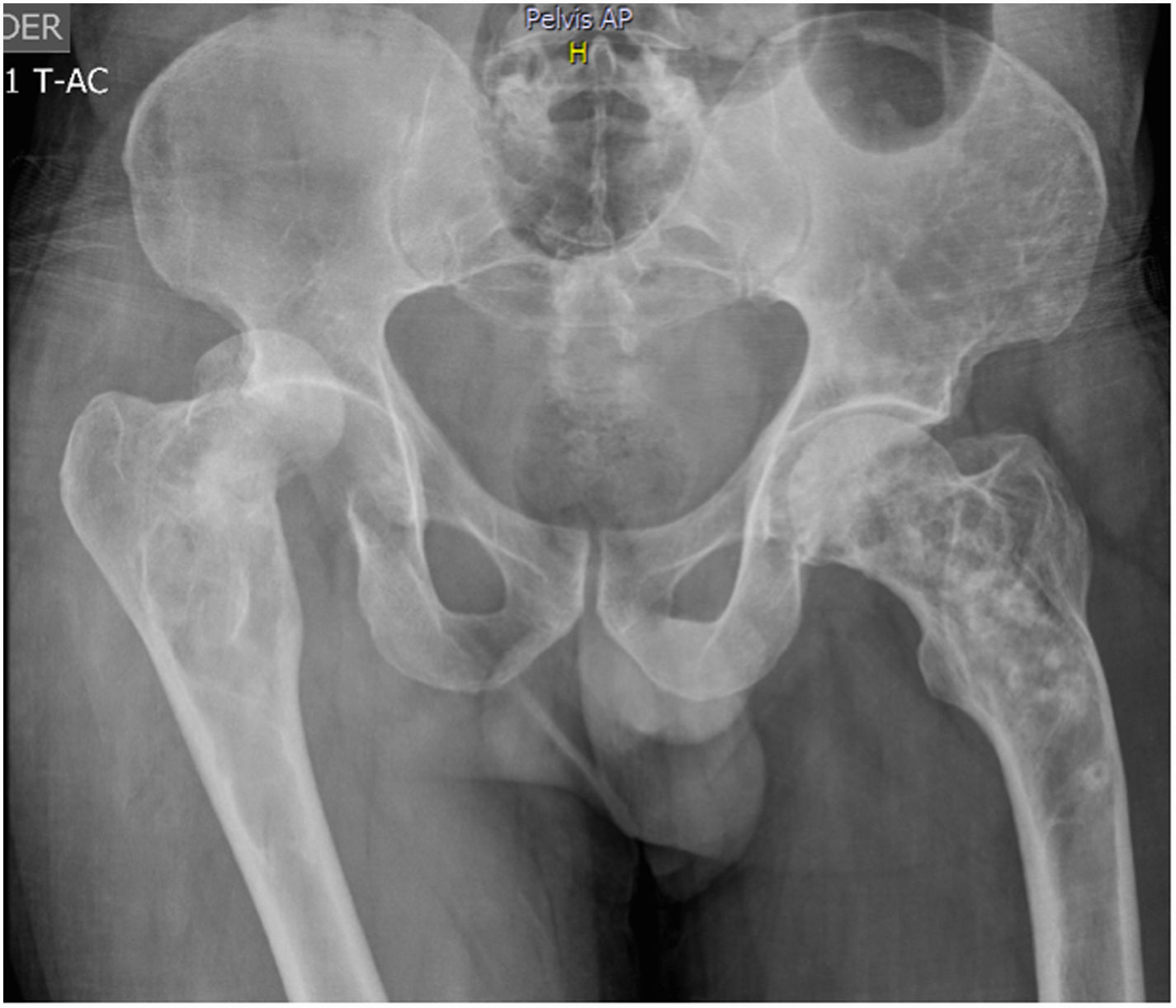
Paciente masculino de 51 años, de ascendencia latina, sin antecedentes patológicos personales ni familiares, previamente asintomático, que consulta tras luxación traumática de cadera derecha. Incidentalmente se encuentra en radiografías simples: presencia de lesiones osteolíticas y osteoblásticas alternantes asimétricas en pelvis - alerón izquierdo, fémur y en columna vertebral con fractura en L3 y L5, en ausencia de hallazgos clínicos o paraclínicos de carcinomas de próstata, tracto gastrointestinal o mieloma. Hemograma, función renal y hepática normales, antígeno prostático normal, calcio corregido 9,7; fósforo 3,3; fosfatasa alcalina 74, 25(OH) vitamina D normal, tomografía toracoabdominal por demás normal. Se hace diagnóstico de EP de hueso poliostótica en estadioIII con compromiso de columna, pelvis y fémur, ante lo cual se indica administración de ácido zoledrónico 5mg sin complicaciones. Tras un año de seguimiento el paciente se encuentra asintomático, sin fracturas ni cambios imagenológicos (fig. 1).
Caso 2
Paciente masculino de 50 años de edad, de ascendencia latina, con antecedente de gota, que consulta por coxartralgia crónica derecha asociada a masa en fémur ipsilateral. Se realiza biopsia de la lesión ósea que revela trabéculas óseas engrosadas e irregulares, patrón de mosaico, líneas cementales basofílicas y alteración trabecular normal, sugestiva de EP monostótica. La gammagrafía ósea revela, además, incremento anormal de captación osteogénica en antepié bilateral, de predominio izquierdo, hipercaptación cortical lateral tibial que sugiere incremento resortivo óseo. Paraclínicos adicionales: fosfatasa alcalina, calcio sérico, fósforo sérico, 25(OH) vitamina D, calciuria y fosfaturia en 24h, todos dentro de rangos de normalidad. Se indica ácido zoledrónico en dosis única con resolución del dolor en cadera; y continúa con febuxostat 80mg diarios y colchicina 0,5mg diarios por patología de base.
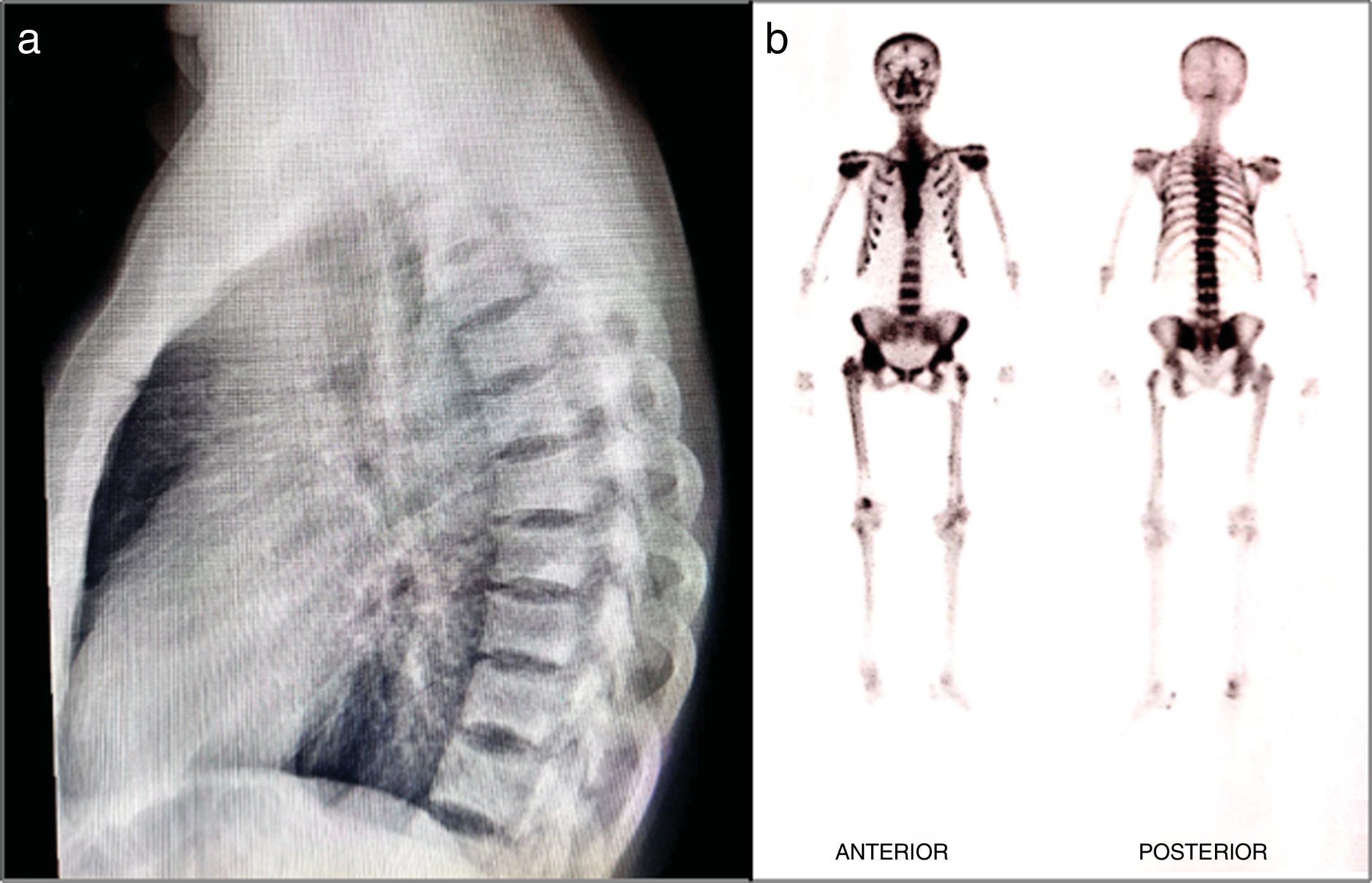
Caso 3Paciente femenina de 48 años, de ascendencia latina, con antecedente de enfermedad renal crónica estadio 5 por nefrolitiasis y reemplazo total de cadera izquierda por artrosis. Consulta por oligoartralgias de grandes articulaciones (hombros, caderas, rodillas) y dolor lumbosacro de patrón mecánico. Al examen físico se evidencia dolor y limitación para la movilidad de hombros, caderas y rodilla izquierda, y dolor a la palpación de región lumbar. En rayos X se observan áreas con aumento de la densidad ósea en cuerpos vertebrales de región dorsolumbar y sacroilíaca y en ambos acetábulos, más cambios de coxartrosis derecha. Por gammagrafía ósea se destaca hipercaptación en cuerpos vertebrales, hombros, caderas, rodillas y tobillos. Densitometría ósea con T-score de columna lumbar 6,8 y en cuello femoral 1,2. Niveles séricos de paratohormona, calcio, fósforo y fosfatasa alcalina, normales. Se hace diagnóstico de EP poliostótica. Por compromiso renal de base, se decide tratamiento con ácido ibandrónico intravenoso trimestral por 5 dosis con adecuada tolerancia, resolución completa del dolor osteoarticular y estabilización de las lesiones óseas tanto por radiografía simple como por gammagrafía ósea (fig. 2).
Revisión de la literatura Rayos X: lesiones osteoblásticas en columna. b) Gammagrafía: lesiones hipercaptantes de predominio en esqueleto axial, hombros y pelvis de un paciente con EP poliostótica III.")
La EP es una enfermedad metabólica del hueso de etiología no esclarecida, que se caracteriza por una fase de resorción aumentada seguida por una fase de osteoformación aberrante. Las primeras publicaciones sobre la enfermedad fueron hechas por Sancerote en 1801, Rullier en 1812, Wilks en 1869 y Czerney en 1873; pero fue solo hasta 1876 cuando el cirujano patólogo británico James Paget describió 5 casos con los hallazgos clínicos e histopatológicos de la enfermedad a la que denominó Osteittis deformans. Describió además una forma de cáncer de mama intraductal y un tipo de adenocarcinoma usualmente no invasivo de la piel que también llevan su nombre. En Colombia, el primer caso de EP fue descrito en 1981 por Sánchez5.
EpidemiologíaEn el mundo, la EP es la segunda enfermedad osteometabólica más común después de la osteoporosis, pero con una distribución geográfica muy heterogénea6. Hay una alta prevalencia en poblaciones del norte de Europa, siendo más elevada en Gran Bretaña, en las ciudades Lancaster, Preston y Bolton (3-5% población mayor de 55 años), y en zonas de emigración de esta población como Australia, Nueva Zelanda o EE.UU. La prevalencia estimada en España es del 1,1 al 1,6%, con zonas de alta prevalencia en áreas geográficas rurales con gran aislamiento y marcada endogamia, como se observa en varias provincias contiguas de la Meseta Central (Sierra de la Cabrera en Madrid, 6,4%; zona de Vitigudino en Salamanca, 5,7%)7,8. La enfermedad es poco frecuente en los países escandinavos y Asia, con una prevalencia menor del 1%. En América del Sur es poco común; sin embargo, se han descrito grandes series de casos en Argentina y Brasil probablemente por una mayor ascendencia blanca europea en estos países. En Colombia la enfermedad es rara, con una frecuencia aproximada de un caso por 1.000.000 habitantes5.
Entre el 5 y el 40% de los pacientes tienen antecedentes familiares de la enfermedad, especialmente en las áreas de alta prevalencia. El riesgo de desarrollar la EP en familiares de primer grado se ha estimado entre 7 y 10 veces mayor que en la población general. También se han descrito familias en las que se hereda como rasgo autosómico dominante con alta penetrancia en la sexta y séptima década de la vida9. Afecta tanto a hombres como a mujeres, siendo en la mayoría de los casos ligeramente más frecuente en los hombres. Es una enfermedad rara antes de los 40 años. Su incidencia aumenta con la edad, estimándose 0,3 casos por 10.000 personas-año en las mujeres y 0,5 casos por 10.000 personas-año en hombres entre los 55 y 59 años. Tasa que aumenta significativamente a partir de los 85 años, en 5,4 para las mujeres y 7,6 para los hombres. La prevalencia del diagnóstico clínico es del 0,3% para hombres y mujeres mayores de 55 años10.
La EP es una enfermedad considerablemente antigua, presente incluso en dinosaurios del Jurásico tardío (150 millones de años), diagnosticada en un ejemplar de Tanzania11.
FisiopatologíaLas lesiones pagéticas óseas evidencian resorción ósea incrementada de carácter osteoclástico, acompañada de fibrosis medular, incremento vascular óseo e incremento de formación ósea, aunque de manera irregular. Histológicamente, reciben el nombre de mosaico, dada la mezcla de tejido óseo y lamelar12. Hay incremento osteoclástico con mayor número de núcleos, así como poseen cuerpos de inclusión nucleares, que pueden recordar partículas virales13. Sin embargo, los cuerpos de inclusión no son específicos para la EP, también pueden estar presentes en patologías como la osteopetrosis14, picnodisostosis15, y la oxalosis hereditaria16.
El rápido recambio óseo en la enfermedad lleva a la producción de hueso, sin embargo, con una arquitectura desordenada, pobre fuerza mecánica y alto riesgo de deformidad y fractura patológica. La enfermedad está gobernada por la acción osteoclástica predominante, por una alta sensibilidad a factores que estimulan la resorción ósea como la 1,25 dihidroxivitamina D, y el ligando de la NFkB (factor nuclear kB), también conocido como RANKL17,18. Dicha asociación, particularmente con RANKL, permite considerar que los osteoclastos presentan un defecto en su apoptosis, lo cual prolonga su supervivencia, pero no ha podido demostrarse aún de manera efectiva. Los cultivos de hueso en pacientes con EP comparados con pacientes sanos muestran una alta sensibilidad para promoción osteoclástica19, así como expresión incrementada de RANKL20, con expresión adicionalmente de IL-1, IL-6, Dickkop-1, que pueden contribuir además a los cambios focales de recambio óseo, característicos de la enfermedad21.
La causa de la EP aún no está completamente dilucidada, pero se han denotado factores genéticos como un claro predisponente para su desarrollo, encontrando que al menos el 15% de los casos tienen trasfondo positivo familiar22,23, y el riesgo de desarrollar la enfermedad se incrementa entre 7 y 10 veces en familiares de primer grado de pacientes, comparados con la población general24. Estudios en muchas familias han permitido identificar que se trata de una enfermedad autosómica dominante, con alta prevalencia en la sexta o séptima década de la vida25-27; se han asociado algunas mutaciones en relación con el desarrollo de la entidad o síndromes relacionados28; el más importante para el desarrollo del Paget clásico es el Sequestosoma 1 (SQSTM1), el cual codifica la p62 (proteína «scaffold»), relacionada con la señalización del NFkB29. La alteración del dominio asociado a la ubiquitina del SQSTM1 se ha reportado como el causal del 20-50% de los casos familiares, y del 5,2% de los casos aislados esporádicos24,30-32. Dicha mutación permite, comparativamente a los que no la poseen, desarrollar una enfermedad más extensa, y con una prevalencia de entre el 79 y 100% en la séptima década de la vida. Otro candidato genético ligado al desarrollo de la EP está en los cromosomas 5q31 y 10p13, aunque los genes relacionados con la enfermedad aún no han sido encontrados25,26.
El mecanismo responsable del desarrollo de la enfermedad, tampoco es claro. Se consideran factores como el estrés mecánico, posible causal de la distribución de las lesiones óseas y, por tanto, su predominancia de presentación en el esqueleto axial y miembros inferiores33. Algunos factores ambientales se han descrito como probables disparadores de la EP. En estudios, observaciones e hipótesis se incluyen: dieta baja en consumo de calcio y vitamina D durante la infancia34,35, exposición ocupacional a toxinas36, infecciones zoonóticas37 y virales38, dentro de ellas la más estudiada es la infección por paramixovirus, particularmente el virus canino distemper y el virus del sarampión32,39,40, dado el aspecto de cuerpos de inclusión nucleares que recuerda la presencia de nucleocápsides virales13, aunque los resultados han sido conflictivos y poco concluyentes41-44. Se ha demostrado el efecto in vitro en el incremento osteoclástico y recambios óseos, aún sin correlación in vivo45.
Manifestaciones clínicas y complicacionesLas manifestaciones clínicas de la enfermedad son muy variables y van desde un curso asintomático hasta cuadros de dolor óseo y complicaciones como sordera, fracturas por deformidad y predisposición a neoplasias. La presentación asintomática es la más común y por esta razón la mayoría de los diagnósticos son incidentales5.
Dolor óseoEl dolor es el síntoma más frecuente, y predomina en miembros inferiores (región lumbosacra, caderas y piernas). La causa del dolor no está claramente dilucidada pero puede deberse a una distensión del periostio en los sitios de formación ósea que comprime las terminaciones nerviosas. Algunos pacientes que evolucionan a la fase osteolítica también se quejan de dolor y otros a pesar de una gran formación ósea no lo manifiestan, lo cual hace de este síntoma un elemento frecuente pero de etiología variable o desconocida. En general el dolor mejora con la movilidad y empeora con el reposo, a menos que exista osteoartrosis secundaria yuxtaarticular a las lesiones de Paget. A su vez el dolor puede ser secundario a complicaciones como fracturas patológicas, fisuras o deformidad, con los desbalances osteomusculares y mecánicos secundarios5.
Los huesos más comúnmente afectados son: pelvis, fémur, columna vertebral, cráneo y tibia, pero puede existir compromiso del esternón, clavículas y costillas. Las lesiones que se manifiestan al inicio se mantienen en el tiempo, y es inusual en el seguimiento la aparición de nuevas lesiones, sin embargo, cuando el hueso afectado es trasplantado puede llegar a desarrollar nuevas lesiones en el receptor. En los huesos largos las lesiones inician en la región proximal de la epífisis y avanzan a lo largo de su eje a una tasa de 8mm/año. El punto máximo del avance de la lesión se observa como una cuña lítica en forma de V que refleja la resorción osteoclástica y aumenta el riesgo de fractura. El mismo fenómeno se observa en el cráneo como una osteoporosis circunscrita46. Después de la fase de resorción ósea, continúa la fase de formación en la cual puede notarse un aumento en el diámetro anteroposterior del cráneo, pronunciado a nivel frontal y occipital. Es infrecuente el compromiso de huesos de la cara. Puede haber compromiso dental con obliteración de la lámina dura y la formación de un cemento óseo en la raíz del nervio y puede existir un fenómeno de anquilosis5.
Complicaciones neurológicasCuando en el cráneo se compromete la cápsula ótica puede aparecer hipoacusia neurosensorial, los pacientes pueden consultar por tinnitus o vértigo5,46. La mielopatía como tal es rara y puede ocurrir por robo vascular más que por compresión.
FracturasPueden presentarse en la fase lítica por desmineralización o en la fase mixta por depósitos de matriz ósea no laminar; los sitios más frecuentes son la tibia y el fémur.
DeformidadPor la fragilidad y pérdida de la arquitectura ósea normal se produce un arqueamiento en los huesos largos, tercio superior de la tibia, fémur, y como consecuencia coxa vara, protrusión del acetábulo, coxa y gonartrosis secundarias.
Transformación maligna de la enfermedad de PagetEsta es una complicación poco frecuente pero severa. En la mayoría de los casos el tumor es un osteosarcoma, pero hay un subgrupo en el que se desarrollan tumores de células gigantes. Este último es un síndrome familiar y se relaciona con mutaciones en el gen ZNF687. Este gen codifica para una proteína C2H2 con anillos de zinc, que es parte de un complejo de regulación transcripcional que se expresa en la mayoría de los tejidos, incluido el hueso. La enfermedad en estos pacientes es usualmente poliostótica y de inicio temprano. Los tumores son multifocales y aparecen luego de 10 años de enfermedad, pero el intervalo puede variar de 3 a 30 años. Esta alteración es más prevalente en el sur de Italia, en quienes se ha encontrado la mutación46.
DiagnósticoEl diagnóstico es fundamentalmente radiológico y frecuentemente incidental como consecuencia del curso asintomático de la mayoría de los casos. La radiografía simple evidencia engrosamiento de las corticales, osteoporosis circunscrita, imágenes escleróticas o líticas alternantes, pérdida de la diferenciación corticomedular, aumento del tamaño del hueso; y también permite evaluar la presencia de complicaciones como deformidades y fracturas47. En el estudio de nuevas áreas de dolor en las que la radiografía simple no es concluyente, puede usarse tomografía computarizada, PET/TC con radiotrazadores como 18F-fluoruro de sodio, por ejemplo, o resonancia48. La gammagrafía ósea tiene una especificidad limitada para el diagnóstico, y aunque puede usarse para valorar la extensión de la enfermedad para lo cual es muy sensible, el identificar compromiso pélvico o vertebral por rayos X ya es determinante de tratamiento antirresortivo, contexto en el cual se podría obviar dicha prueba.
Según los hallazgos radiográficos de la enfermedad, esta se categoriza en «activa» e «inactiva». La primera se subdivide en temprana-etapa I u osteolítica, en la que se afectan cráneo y huesos largos con osteoporosis circunscrita y osteoporosis subcondral con radiolucencia que avanza en forma de cuña; temprana-etapa II o osteolítica/osteoesclerótica/ambas, en la que se afectan cráneo, huesos largos y pelvis con los cambios descritos en etapa I más radiolucencias y radiodensidades en parches que comprometen diáfisis, epífisis y metáfisis. La categoría inactiva, tardía, etapa III u osteoesclerótica incluye afección de cráneo, huesos largos, pelvis y columna; en estos casos predomina la apariencia en motas de algodón, engrosamiento de la bóveda craneana y de los huesos de la pelvis con radiodensidad focal o difusa, cuerpos vertebrales en «marco de cuadro» o en «marfil», predilección epifisiaria, engrosamiento trabecular, ensanchamiento y deformidad del hueso5.
Desde el punto de vista serológico, el hallazgo incidental de fosfatasa alcalina total (FAT) elevada, en relación con niveles elevados de gamma glutamil transpeptidasa (GGT), indican patología hepatobiliar. Por el contrario, niveles normales de GGT en una persona adulta no embarazada probablemente son de etiología ósea. La FAT está elevada en el 85-95% de los pacientes con EP sin tratamiento, se correlaciona con la actividad de la enfermedad, y por ello puede usarse como biomarcador de seguimiento. Niveles normales de fosfatasa alcalina se presentan en enfermedad monostótica, pocos huesos afectados o enfermedad metabólicamente inactiva49.
La medición de otros marcadores de formación ósea como osteocalcina y péptidos de procolágeno tipo I y de marcadores de resorción ósea, que incluyen las piridinolinas, hidroxiprolina y colágenos reticulados, no ofrece ninguna información adicional, no son específicos y el diagnóstico se establece mejor mediante imágenes1,50. Sin embargo, algunos autores plantean que el propéptido N-procolágeno-1 parece funcionar mejor, en términos de elevarse por encima de lo normal incluso si la enfermedad es de extensión limitada51. Por muchos años se usó la medición de hidroxiprolina urinaria; sin embargo, entró en desuso por lo laborioso de la prueba y por limitaciones de sensibilidad y especificidad.
En muy pocas ocasiones se requiere la toma de una biopsia ósea para establecer el diagnóstico, aunque puede ser útil para diferenciarlo de metástasis osteoblásticas u osteosarcomas si la clínica lo sugiere. Histológicamente la microestructura del hueso es altamente anormal en EP. Los osteoclastos aumentan en número, tamaño y nuclearidad, y presentan inclusiones nucleares. Inicialmente se pensaba que estas inclusiones eran paramixovirus, pero más recientemente se ha sugerido que son agregados de proteínas no degradadas debido a defectos en la vía de la autofagia. La formación de hueso también se incrementa, en respuesta al aumento en la resorción ósea. Sin embargo, el hueso recién formado es anormal y está depositado de manera caótica, lo que da como resultado un hueso mecánicamente débil. Otras características histológicas de la EP activa incluyen aumento de la vascularización y fibrosis de la médula52.
Análisis histológicos como la histomorfometría de vértebras y pelvis se ha usado en estudios de investigación y confirman mayor grosor trabecular, alto recambio óseo con un aumento significativo en la resorción ósea y los índices de formación ósea (número trabecular, volumen osteoide y superficie osteoide, número de osteoblastos y superficie de los osteoclastos), y un aumento del volumen óseo. Sin embargo, se afirma que hay una mejor correlación entre la formación de hueso y la resorción ósea que en la osteoporosis10.
Diagnósticos diferenciales: enfermedades de carácter congénito, degenerativo, infeccioso (osteomielitis crónica), metabólico (raquitismo, osteomalacia, hiperparatiroidismo, hipertiroidismo, osteodistrofia renal), traumático o maligno (primario por osteosarcomas o metastásico especialmente por cáncer de próstata y mieloma; enfermedades como la displasia fibrosa)46. En estos casos será valiosa la información por historia clínica de síntomas asociados y hallazgos alterados de laboratorio.
TratamientoEl tratamiento de la EP actualmente genera ciertas controversias. Se han planteado 3 razones principales para llevar a cabo un manejo farmacológico. La primera, desde el punto de vista fisiopatológico, busca normalizar tanto como sea posible el recambio óseo53. La segunda razón es clínica, para el control del dolor; y la tercera tiene como objetivo tratar las complicaciones. Estos 3 objetivos básicos pueden lograrse en general con el uso de los bifosfonatos, los cuales han demostrado ser eficaces en la disminución del recambio óseo y en el control del dolor, pero al parecer son menos efectivos en prevenir varias de las complicaciones de esta enfermedad.
El uso de analgésicos, iniciando por el paracetamol hasta otros analgésicos más potentes, permite en muchos casos el control adecuado del dolor generado por la EP. Sin embargo, la seguridad de los analgésicos a largo plazo y la falta de efecto sobre la fisiopatología de la enfermedad conllevan buscar otro tipo de tratamientos. La calcitonina, históricamente, fue uno de los primeros fármacos usados para el control de la EP, debido a su actividad sobre el recambio óseo54. No obstante, su corto efecto de acción generó que prontamente fuera reemplazada por los bifosfonatos y hoy en día su uso es cada vez menos frecuente55.
Diferentes bifosfonatos han sido utilizados para el tratamiento de esta enfermedad. En la actualidad se cuenta con bifosfonatos de mayor potencia y duración de su efecto de acción. Han demostrado en diversos estudios (algunos de ellos aleatorizados y controlados), no solamente el control del dolor óseo, sino también otros efectos como disminuir las lesiones líticas, disminuir el recambio óseo y restaurar la calidad del hueso56. La discusión se ha centrado en el tiempo de uso y en su capacidad para evitar otras complicaciones como la sordera o neurocompresiones. En cuanto al tiempo de uso, diferentes esquemas de tratamiento han sido utilizados, dependiendo también del tipo de bifosfonato usado. Sin embargo, el riesgo de recaídas tras su suspensión ha llevado a la búsqueda de protocolos de monitorización y tratamiento más adecuados. El pamidronato, luego de una dosis inicial de 90mg intravenosos (divididos en sesiones de 30mg por día), puede tener un efecto hasta por 12 meses aproximadamente. Otros bifosfonatos como el alendronato (no avalado en varios países europeos) o el risedronato pueden tener una duración de su efecto hasta por 5 años, pero con el uso de dosis diarias durante varios meses. Otros medicamentos de este tipo, como el etidronato, el tiludronato, etc., son menos usados. En la actualidad el zoledronato es considerado el bifosfonato más potente y con mayor duración de su efecto, pudiendo lograr respuesta clínica y sobre el recambio óseo por más de 6 años, luego de una sola dosis intravenosa1.
Cuando se utilizan los niveles séricos de FAT para monitorizar el efecto de estos medicamentos, se ha demostrado con el zoledronato una reducción del 75% de esta enzima en el 98% de los pacientes durante 6 meses. La recaída (definida como una elevación de la FAT superior al 20% del valor pretratamiento) solo ocurrió en menos del 1% de los pacientes a 6 años y medio de observación57,58. Respecto a la capacidad de los bifosfonatos para evitar otras complicaciones descritas, el estudio PRISM no evidenció una diferencia significativa entre el uso de bifosfonatos y el uso de analgésicos, mostrando que la tasa de complicaciones a largo plazo era similar, sin notarse una mayor reducción con el uso de bifosfonatos. Sin embargo, es posible que sean también efectivos en una rara complicación por paraparesia como consecuencia del compromiso vertebral59.
Recientemente, se ha planteado el uso del denosumab, el cual teóricamente podría ser una alternativa atractiva de tratamiento, pero hasta ahora su uso ha sido anecdótico, aunque con resultados aparentemente alentadores54,60.
Finalmente, para el tratamiento de complicaciones mecánicas asociadas a las deformidades óseas de la EP, se ha planteado la cirugía ortopédica. Sin embargo, por la mayor vascularización ósea que se presenta en esta enfermedad, se incrementa el riesgo de hemorragias durante este tipo de intervenciones. Por esta razón, se han utilizado esquemas de pretratamiento con bifosfonatos, con el fin de reducir este riesgo de sangrado o de pérdida de prótesis debido al recambio óseo acelerado. De todas maneras, el tratamiento médico previo obliga necesariamente a retrasar algunos meses el acto quirúrgico61. Otras complicaciones como la sordera, compresiones medulares por lesiones vertebrales o la falla cardíaca han recibido también tratamiento con bifosfonatos con respuestas poco favorables.
DiscusiónLa EP es una enfermedad osteometabólica muy prevalente en Europa y en áreas de emigración de esta población. Sin embargo, es muy infrecuente en Asia, Medio Oriente y África. En América Latina la distribución es heterogénea y, concretamente, en Colombia se siguen reportando casos esporádicos. Sumando los 3 casos que en el presente artículo se reportan, a los descritos por Sánchez et al.2-4, se tienen 10 casos, de los cuales todos fueron mayores de 40 años; edad a partir de la cual se han detectado los pacientes en la literatura mundial. Pese a que afecta a ambos géneros de forma similar, el 70% de los casos que tenemos reportados son mujeres, sin que se logre determinar si realmente hay un factor hormonal, cromosómico, entre otros, que favorezca dicha diferencia de la misma forma como sucede con la osteoporosis. De forma paralela, los mecanismos responsables del desarrollo de la enfermedad, como el estrés mecánico, factores infecciosos y nutricionales, no tienen francas diferencias entre los géneros, en nuestro medio. Ninguno de los 3 casos descritos tiene antecedentes familiares reconocidos con EP, lo que sugeriría que más allá de factores genéticos, en nuestro medio deben contemplarse factores ambientales múltiples, no claramente conocidos, como posibles inductores de que aparezca la patología.
Las manifestaciones clínicas fueron las descritas a escala mundial sin que se tuviesen complicaciones neurológicas, fracturas, deformidades o transformaciones malignas. Pese a que algunos pacientes tuvieron estudios de extensión, a todos se les hizo el diagnóstico por hallazgos típicos en radiografías simples. Seis de ellos con enfermedad poliostótica todos en categoría inactiva, tardía o etapa III, con cambios especialmente escleróticos como corresponde. Tres pacientes tuvieron FAT normal; esto último es posible especialmente en casos de inactividad, como en dichos casos46.
Las razones de tratamiento fueron normalizar el recambio óseo, modular el dolor y evitar complicaciones por compresión neurológica en aquellos con compromiso vertebral ante el planteamiento en la literatura de que puede ofrecer minimización del riesgo de paraparesia59. La calcitonina, históricamente, fue uno de los primeros fármacos usados para el control de la EP, pero debido a su corto efecto de acción fue reemplazada por los bifosfonatos, en especial el ácido zoledrónico en unidosis, pues permite minimizar las recaídas de la enfermedad por periodos más prolongados56. En los casos recientemente reportados se usaron bifosfonatos.
ConclusionesLa EP es una enfermedad osteometabólica crónica, con distribución geográfica heterogénea. Rara en nuestro medio, que se presenta asintomática o con dolor óseo tanto en hombres como en mujeres mayores de 40 años. El diagnóstico es eminentemente radiográfico y el tratamiento se basa en el uso de bifosfonatos, especialmente cuando se busca control del dolor, y aún no es claro que realmente tenga impacto en la disminución del riesgo de complicaciones, especialmente de compresión neurológica cuando hay compromiso vertebral.
Conflicto de interesesNo hay conflicto de interés de ninguno de los autores.
