




Idiopathic inflammatory myopathies (IIM) are a heterogeneous group of diseases characterised by skeletal muscle involvement, manifested by weakness and inflammatory signs in the muscle biopsy. The objective of this article is to describe the clinical, laboratory, and treatment features of a cohort of patients with IIM in southwest Colombia.
MethodsA retrospective review was conducted on the medical records of patients diagnosed with IIM treated at a fourth-level complexity hospital in Cali, Colombia, from 2011 to 2017. Demographic, clinical, serological, and treatment data were collected.
ResultsA total of 72 patients with IIM were identified, mostly women (n = 54, 75%). The mean age at onset of symptoms was 37.11 ± 19.18 years. The main subtypes of IIM were dermatomyositis (DM) and polymyositis, occurring in 35 patients (48.6%) and 25 patients (34.7%), respectively. Twenty-eight patients (38.8%) had associated autoimmune disease, with systemic lupus erythematosus being the most frequent in 7 (9.72%) patients. Muscle biopsy was performed in 25 patients (34.7%), while 28 (38.8%) had positive antinuclear antibodies. The median creatine phosphokinase was 877.5 mg/dL (163.5–4358.3). Sixty-seven patients (93.1%) were treated with glucocorticoids, and 18 (25%) patients were treated with rituximab (RTX) as monotherapy or combined with another immunosuppressant drug.
ConclusionsDM is the most frequent subtype of IIM, being common in women and occurring in the fourth decade of life. The most used treatments were glucocorticoids, followed by RTX monotherapy, or combined with other immunosuppressants.
Las miopatías inflamatorias idiopáticas (MII) constituyen un grupo heterogéneo de enfermedades que comprometen la musculatura esquelética y se manifiestan por debilidad y signos inflamatorios en la biopsia muscular. El objetivo de este estudio es hacer una caracterización epidemiológica de una cohorte de pacientes con MII en una población del suroccidente colombiano.
MetodologíaDe forma retrospectiva, se revisaron las historias clínicas de pacientes con diagnóstico de MII que fueron tratados en un hospital de cuarto nivel de complejidad en Cali, Colombia, entre el 2011 y el 2017. Se recolectaron variables demográficas, clínicas, serológicas y de tratamiento.
ResultadosSe identificaron 72 pacientes con MII, mayoritariamente mujeres (n = 54, 75%). La media de edad al inicio de los síntomas fue de 37,11 ± 19,18 años. Las principales MII fueron dermatomiositis (DM) y polimiositis, las cuales se presentaron en 35 (48,6%) y 25 pacientes (34,7%), respectivamente. Veintiocho pacientes (38,8%) presentaban enfermedad autoinmune asociada, siendo el lupus eritematoso sistémico la más frecuente, al presentarse en 7 (9,72%) pacientes. La biopsia de músculo se realizó en 25 pacientes (34,7%), mientras que 28 (38,8%) tenían anticuerpos antinucleares positivos. La mediana de la creatinfosfoquinasa fue de 877,5 mg/dl (163,5-4.358,3). Sesenta y siete pacientes (93.1%) fueron tratados con glucocorticoides y 18 (25%) con rituximab (RTX) como monoterapia o combinado con otro fármaco inmunosupresor.
ConclusionesLa DM es la condición clínica más frecuente, es común en mujeres y se presenta en la cuarta década de la vida. Los tratamientos con los que más se obtuvo mejoría clínica fueron los glucocorticoides, seguidos del RTX en monoterapia o combinado con otros inmunosupresores.
Idiopathic inflammatory myopathies (IIM) are a heterogenous group of rare diseases, characterized by multisystem involvement resulting in non-suppurative inflammation of the skeletal muscles which may be accompanied by organic skin, joints, lung, GI tract and heart disorders.1 These conditions may severely deteriorate the quality of life.2 Their incidence and prevalence ranges between 1.16 and 19 million per year and between 2.4 to 33.8 cases per every 100,000 inhabitants, respectively, depending on the geographical area, the research methods used, and the classification criteria followed.3
The conditions included in the group of IIM are dermatomyositis (DM), polymyositis (PM), immune-mediated necrotizing myopathy (IMNM) and inclusion-body myositis (IBM).1 The primary symptom is muscle weakness, though there may an organ-specific dysfunction resulting in a particular clinical syndrome. The serology of muscle inflammation is expressed as elevation of creatine phosphokinase (CPK), aldolase, lactic dehydrogenase (LDH) and transaminases. The diagnostic tools to show myopathic involvement include electromyography and magnetic resonance imaging which also helps to guide the biopsy. Biopsy is important because the specific histopathological findings enable the differentiation of the various clinical subtypes of IIM.4
The most widely used classification criteria for IIM are the Bohan and Peter5 criteria and Tanimoto et al.,6 which are based on clinical, histopathological and neurophysiological findings, in combination with elevated levels of muscle enzymes, and specific autoantibodies (only in Tanimoto et al.). However, no other useful findings are included to differentiate the subtypes of patients with myositis, such as MRI and immunohistochemistry characterization, which includes the major histocompatibility complex (MHC) molecules in the muscle fibers and subtypes of inflammatory cells involved.4
Currently there are few descriptions of patients with IIM in Colombia; however, this is the largest series published to this date. This study describes the clinical, serological, and treatment characteristics of patients with a diagnosis of IIM in a fourth-level hospital in Cali, Colombia.
MethodsPatientsA retrospective review was conducted of the medical records of patients with a diagnosis of IIM, according to codes ICD-10, treated at Fundación Valle del Lili, a fourth-level hospital in Cali, Colombia, between January 1st and December 31st, 2017. The patients selected met the Bohan and Peter5 classification criteria for IIM; patients under 18 years old and patients with a diagnosis of myopathy due to toxic, metabolic, infectious, or neuromuscular causes were excluded.
The clinical and laboratory characteristics were recorded at the time of diagnosis and during the follow-up. Any associated auto-immune diseases were defined in accordance with the classification criteria for the respective diseases (rheumatoid arthritis, systemic lupus erythematous systemic sclerosis, Sjögren’s syndrome, vasculitis). Patients who failed to respond to 2 immunomodulators or immunosuppressors when administered at their maximum dose for 3 months (either individually or simultaneously), were considered refractory to treatment. Remission of the disease was defined as the normalization of the CPK values, and improved muscle weakness for a continuous period of 6 months or longer. This study was approved by the ethics committee of our institution.
Statistical analysisA statistical descriptive analysis was conducted. The continuous variables were expressed as averages and standard deviation or median and interquartile ranges, and were analyzed using the t test or Mann-Whitney U test, in accordance with normal distribution. The data were analyzed using STATA 12.1.
Results270 patients with a diagnosis of myopathy were seen between 2011-2017. After reviewing the medical records, 198 patients were ruled out, in accordance with the exclusion criteria. A total of 72 patients with IIM was identified, of which 18 (25%) were males and 54 (75%) were females; the female prevalence was 3:1. The mean age of the patients at the onset of symptoms was 37.11±19.18 years. With regards to the IIM distribution, 35 patients (48.6%) had DM, 25 (34.7%) PM, 8 (11.1%) juvenile dermatomyositis, 2 (2.7%) IBM, 1 (1.3%) IMNM, and 1 (1.3%) non-specified inflammatory myopathy.
In terms of the frequency of symptoms and signs, most of the patients included (72%) experienced muscle weakness and 17 (23.6%) had dysphagia. The dermatological manifestations included Gottron papules in 21 patients (29.7%), Gottron sign in 9 (12.5%), heliotrope rash in 10 (13.8%), and the shawl sign in 3 (4.1%). In terms of comorbidities, 25 patients (34,7%) had associated autoimmune disease. Systemic Lupus Erythematous (SLE) is the most frequent condition and it was present in 7 patients (9.7%), followed by scleroderma, in 6 (8.3%); rheumatoid arthritis in 5 (6.94%); Sjögren’s syndrome in 3 (4.2%); central nervous system vasculitis, cutaneous leukocytoclastic vasculitis, autoimmune polyglandular syndrome, and pyoderma gangrenosum, in one patient (1.4%) each. Additionally, 39 patients (54.17%) had non-autoimmune comorbidities. However, 19 (26.38%) presented more than 2 non-autoimmune concomitant diseases, of which the most frequent were: high blood pressure in 11 cases (15.28%), thyroid disease in 10 (13.89%) and diabetes mellitus in 6 (8.33%).
Neoplasms were identified in 3 patients: 2 hematological malignancies (myeloproliferative and lymphoproliferative syndrome) and one thyroid carcinoma. Interstitial pulmonary involvement was present in 14 patients (19%), of which 5 had the ground-glass pattern (35.7%), as well as honeycombing. One of these 5 patients had overlapping scleroderma and another one had the combination of both patterns. A restrictive pulmonary pattern was described in the remaining cases. In these cases, there was overlapping scleroderma and one case with scoliosis. Likewise, of the group of patients with pulmonary involvement, the majority (n=6, 42.8%) achieved clinical remission at the last rheumatology control visit, while one patient (7.1%) required permanent oxygen support at home; another one (7.1%) required pulmonary rehabilitation therapy and the rest of the patients presented with some level of persistent clinical compromise, such as dyspnea.
25 patients (34.7%) underwent muscle biopsy, which helped to confirm the diagnosis. In the remaining 47 patients (65.3%) the diagnosis was supported by electromyography with myotatic pattern, MRI with evidence of muscle edema and the clinical opinion of the rheumatologist. With regards to laboratory tests, 28 patients (38.8%) had positive antinuclear antibodies (ANA) (defined as a titer ≥1:160), with the mottled pattern being the most frequent, occurring in 14 patients (19.4%). The anti-Jo1 was positive in only patient. Finally, the erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) were high in 33 (45.8%) and 26 (36%) patients, respectively. Table 1 summarizes the clinical and serological characteristics of the patients with IIM.
Clinical, demographic and serologic characteristics.
| Characteristics | n (%)=72 |
|---|---|
| Females, n (%) | 54 (75%) |
| Age at onset of symptoms, years* | 37.11±19.18 |
| Type of myopathy, n (%) | |
| Dermatomyositis | 35 (48.61) |
| Polymyositis | 25 (35%) |
| Juvenile dermatomyositis | 8 (11.11) |
| Inclusion-body myopathy | 2 (2.78 |
| Necrotizing myopathy | 1 (1.39) |
| Unspecified inflammatory myopathy | 1 (1.39) |
| Associated autoimmune disease, n (%) | |
| Systemic Lupus Erythematous | 7 (9.72) |
| Scleroderma | 6 (8.33) |
| Rheumatoid arthritis | 5 (6.94) |
| Sjögren’s syndrome | 3 (4.17) |
| Other | 4 (5.55) |
| Clinical characteristics, n (%) | |
| Symmetrical muscle weakness | 52 (72%) |
| Dysphagia | 17 (23.61) |
| Heliotrope rash | 10 (13.89) |
| Gottron papules | 21 (29.17) |
| Gottron sign | 9 (12.5) |
| Shawl sign | 3 (4.17) |
| Serologic characteristics, n (%) | |
| ANA positive | 28 (38.8%) |
| Mottled | 14 (19.44) |
| Homogeneous | 8 (11.11) |
| Granular | 3 (4.17) |
| Nucleolar | 3 (4.17) |
| CPK at diagnosis (mg/dL)** | 877.5 (163.5−4.358.3) |
| Aldolase at diagnosis** | 6.7 (0−27.3) |
| AST at diagnosis** | 57.35 (29.8−177.7) |
| ALT at diagnosis** | 47.5 (26.2−125.3) |
| LDH at diagnosis** | 376 (270−618.5) |
| Diagnosis based on biopsy | 25 (34.72) |
| Bohan and Peter Criteria *** | |
| Definitive | 22 (30.5) |
| Probable | 25 (34.7) |
| Possible | 18 (25) |
| Disease remission | 34 (47.22) |
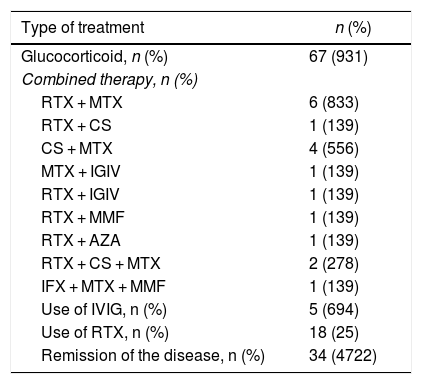
As to the treatment received, most patients (n=67, 93.1%) were treated with glucocorticoids. The immunomodulatory therapy represents a glucocorticoid-sparing option. In general, the patients received different conventional immunomodulatory therapies over the course of the disease, such as methotrexate in 34 patients (47.2%) and azathioprine in 23 (31.9%). However, a clinical response was observed in 18 rituximab (RTX)-treated patients (25%), either as monotherapy or as combined therapy with another immunosuppressant agent (methotrexate, 6 (8.3%); ciclosporin, one (1.3%); intravenous immunoglobulin, one (1.3%); azathioprine, one (1.3%); and mycophenolate mofetil, one (1.3%). Thirteen of the RTX-treated patients responded to treatment, in a mean time of 60 days (IQR: 36–141) following the administration of the first dose of the drug, showing an objective improvement in muscle strength and a decline in muscle enzymes. Before the administration of RTX, the mean CPK was 687 U/L (IQR: 205−3,368), and after its administration, considering the report closest to the muscle strength evaluation, the median CPK was 167 U/L (IQR: 81.5–506.5).
The most frequently used combined therapy after RTX-methotrexate was cyclosporine - methotrexate, which was used in 4 patients (5.56%), while intravenous immunoglobulin was used in 5 patients (5.64%). While RTX was widely used, either as monotherapy or in combination with another drug, it was not the first line of therapy in any of the cases, in contrast with glucocorticoids that were initially used in all patients. 34 patients (47.2%) achieved remission of the disease (Table 2).
Treatment.
| Type of treatment | n (%) |
|---|---|
| Glucocorticoid, n (%) | 67 (931) |
| Combined therapy, n (%) | |
| RTX + MTX | 6 (833) |
| RTX + CS | 1 (139) |
| CS + MTX | 4 (556) |
| MTX + IGIV | 1 (139) |
| RTX + IGIV | 1 (139) |
| RTX + MMF | 1 (139) |
| RTX + AZA | 1 (139) |
| RTX + CS + MTX | 2 (278) |
| IFX + MTX + MMF | 1 (139) |
| Use of IVIG, n (%) | 5 (694) |
| Use of RTX, n (%) | 18 (25) |
| Remission of the disease, n (%) | 34 (4722) |
AZA: azathioprine; CS: ciclosporin; IFX: infliximab; IVIG: intravenous immunoglobulin; MMF: mycophenolate mofetil; MTX: methotrexate; RTX: rituximab.
This is the largest registry of IIM patients in Colombia. The purpose of the study was to retrospectively analyze the clinical, laboratory and treatment characteristics of patients with IIM in a hospital in Cali, Colombia.
With regards to the IIM subtype, the most frequent condition in practically fifty percent of the patients was DM, in contrast to other studies in which PM was the most common condition.7,8 This finding could be associated with the fact that several patients were reclassified during the diagnosis process as DM, following the histopathological findings compatible with this condition. A muscle biopsy was conducted in 35% of the patients; this represents one additional tool for the accurate diagnosis of the different groups of IIM.9–11
IIMs are associated with different autoimmune diseases and are described with a very variable frequency, ranging from 7% to 60%, according to the different series.12,13 In our population, the association with autoimmune diseases was 35%, primarily with SLE. In a retrospective study conducted in Brazil, with a cohort of 220 patients, the intent was to study the overlapping of syndromes (PM/DM); they found that the most common associations were systemic sclerosis, SLE and RA, with frequencies of 48.4%, 29% and 22.6%, respectively.14 In a cohort of 160 patients with IIM, of those patients with overlapping syndromes (39), 33.3% had scleromyositis, 27.6% RA, 23.1% Sjögren’s syndrome and 12.8% SLE.15
ANA may be positive in around 5.9%–30.8% of the healthy population.16 These are more frequent in women and elderly individuals.17 ANA may also be positive in autoimmune diseases such as RA, Sjögren’s syndrome, systemic sclerosis, mixed connective tissue and IIM disease. In our population, 38.8% of the patients had positive ANA, with the mottled pattern being the most frequent. Moreover, the anti-Jo1 was positive in only one patient. This antibody belongs to the myositis specific antibodies (MSA) and is the most frequent antibody (up to 20%) in IIM. Additionally, it is associated with the antisynthetase syndrome and is a marker for poor prognosis.18 The rest of the panel of specific autoantibodies was not assessed, since those are not available in our institution.
The association between IIM and malignancy was reported back in 1916.19 One of the largest IIM cohorts in Australia included 537 patients with a diagnosis confirmed with biopsy, identifying 116 malignancies in 104 patients, with a stronger association among DM patients.20 A recent meta-analysis described an increased risk for malignancy in patients with PM and DM, with a relative risk (RR) of 1.62 (95% CI 1.19–2.04) and 5.50 (95% CI 4.31–6.70), respectively.21 However, in our study, only 3 patients had neoplastic diseases associated with inflammatory myopathy.
In terms of treatment, there are 3 key pillars: control or elimination of causal factors; the used of immunosuppressive therapy, and rehabilitation. With regards to immunosuppressants, GC are the first line of therapy. In our study, almost all patients received glucocorticoids. Methotrexate, ciclosporin, azathioprine, mycophenolate mofetil, cyclophosphamide and IVIG represent an option for refractory cases, or are used as GC-sparing agents. Moreover, in patients who are refractory to first line therapy, upon ruling out any other underlying condition (such as necrotizing myopathy or inclusion body myositis), the use of biologics such as RTX22 should be considered, which may be administered in combination with another immunomodulator or as monotherapy with GC.
RTX is a monoclonal antibody that binds to the CD20 antigen expressed on the surface of B lymphocytes, resulting in the depletion of positive CD20 in the peripheral blood, within the next 6–9 months.23 It was empirically used in patients who failed to adequately respond to conventional therapy, according to the premise and evidence of circulating antibodies in around 80% of the patients with IIM and of B cells in the perivascular region of the muscles in this group of patients.24–26 In our study, 18 patients received this type of immunomodulatory therapy combined with RTX, with methotrexate being the most widely used drug, together with RTX. The observed remission was around 40%, in contrast to other trials reporting less than 10%.8
The limitations of the study included not being able to conduct muscle biopsies in all patients, considering that it was not part of the regular protocol when patients began to be enrolled. Another limitation is the potential selection bias due to the retrospective nature of the study, and the inclusion of patients classified according to IDC-10.
ConclusionThe most prevalent inflammatory myopathy in this cohort was DM, which is frequent in women and usually develops in the fourth decade of life. Glucocorticoid therapy was the most frequently used. Combined immunosuppressive therapy represents an alternative to control the disease.
Please cite this article as: Santos VA, Aragón CC, Posso-Osorio I, Obando MA, Barrera T, Zamorano L, et al. Caracterización epidemiológica de pacientes con miopatía inflamatoria en un hospital de cuarto nivel en Cali, Colombia. Rev Colomb Reumatol. 2021;28:83–88.
