



Autoimmune pancreatitis is a characteristic manifestation of the spectrum of the disease related to IgG4, a rare autoimmune disorder that presents clinically with obstructive jaundice due to the infiltration of plasma cells and fibrosis in the pancreas. There may be other symptoms in case of involvement of other organs, and in very rare cases there is hematological involvement. We present the case of an adult man with signs of cholestasis secondary to type I autoimmune pancreatitis, with involvement of other organs and associated with thrombocytopenia that improved with systemic corticosteroid-based immunosuppressive treatment, after which the patient showed favorable clinical and analytical evolution over time.
La pancreatitis autoimmune es una manifestación característica del espectro de la enfermedad relacionada con IgG4, trastorno raro de tipo autoinmune que se presenta clínicamente con ictericia obstructiva debido a la infiltración de células plasmáticas y fibrosis en el páncreas; puede presentarse con otra sintomatología en caso de afectación de otros órganos, y en muy raras ocasiones hay compromiso hematológico. Se presenta el caso de un hombre adulto con signos de colestasis secundaria a una pancreatitis autoinmune tipo I, con compromiso de otros órganos y asociada con trombocitopenia que mejoró con el tratamiento inmunosupresor a base de corticoide sistémico, luego del cual se observó una evolución favorable en cuanto a la clínica y analítica en el transcurso del tiempo.
A 60-year-old man, with no known clinical or surgical antecedents, who attended the Emergency Department due to a clinical picture of 8days of evolution characterized by the presence of spontaneous ecchymoses and petechiae in the abdomen that subsequently spread to the rest of the body; 3days before admission he also presented jaundice, pruritus, acholia and choluria. As an accompanying symptom he reported loss of appetite and unquantified weight loss, without presenting abdominal pain or temperature rise.
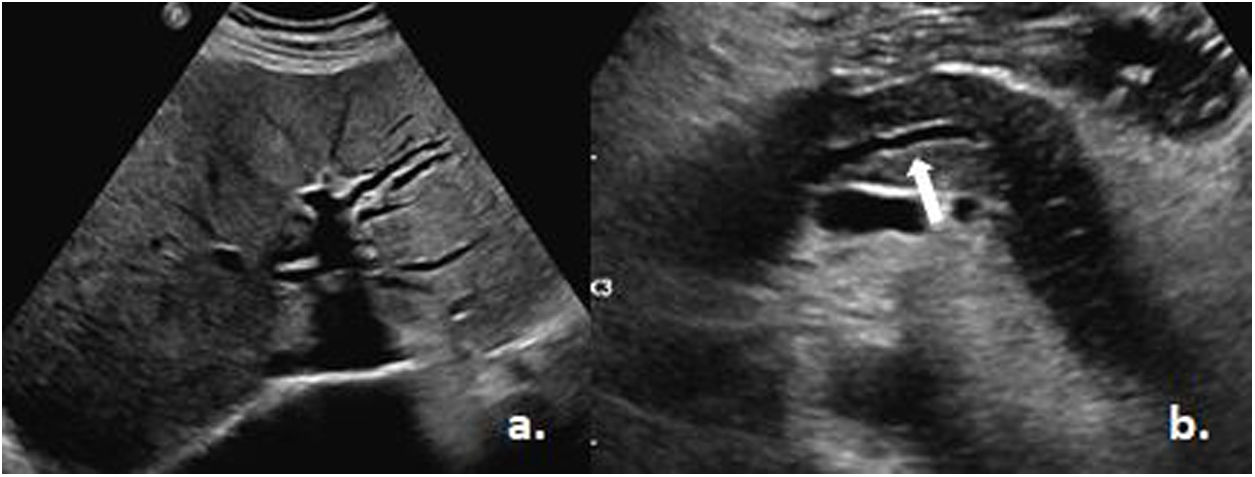
On admission, the patient was awake, lucid and afebrile, with vital signs within normal parameters and generalized jaundice. The paraclinical studies showed leukopenia, lymphopenia and severe thrombocytopenia, as well as an altered liver profile with the presence of hyperbilirubinemia at the expense of direct bilirubin and elevated liver enzymes with a predominant cholestatic pattern (Table 1). An abdominal ultrasound was performed, which did not report obstructive or space-occupying lesions, with marked dilatation of the intra- and extrahepatic bile ducts and the presence of a hypoechoic pancreas of normal size with mild dilatation of the Wirsung duct (Fig. 1).
Biochemical and hematological follow-up.
| Laboratory evolution | Day 0 | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | Day 11 | Day 17 | Day 33 | Day 52 | 1st Year |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total B. | 12.9 | 8.07 | 5.63 | 5.31 | 3.8 | 2.1 | 1.04 | ||||
| Direct B. | 11.36 | 6.93 | 4.86 | 4.51 | 3.07 | 1.87 | 0.73 | ||||
| Indirect B. | 1.57 | 1.14 | 0.77 | 0.8 | 0.73 | 0.23 | 0.31 | ||||
| GGT | 890 | 528 | 321 | 149 | 50 | ||||||
| AP | 540 | 342 | 267 | 200 | 91 | ||||||
| AST | 132 | 51 | 38 | 33 | 17 | 18 | |||||
| ALT | 160 | 106 | 115 | 93 | 39 | 20 | |||||
| Leukocytes | 3800 | 3810 | 3600 | 4590 | 8830 | 7300 | 7100 | 11,010 | 6510 | 9360 | |
| Lymphocytes | 470 | ||||||||||
| Platelets | 6000 | 5000 | 9000 | 24,000 | 82,000 | 136,000 | 193,000 | 186,000 | 171,000 | 278,000 | 193,000 |
| IgG4 | 887 (3−201) | 88.7 | |||||||||
| CA 19-9 | 1747 (0−34) | 866.8 | 16.92 |
Bilirubin value: mg/dl; GGT, AST, ALT: U/l; leukocytes, lymphocytes and platelets: K/uL; IgG4: mg/dl; Ca 19-9: U/mL.
Taken from the database service of the Metropolitan Hospital.
 Dilatation of the biliary tract. b) Dilatation of the Wirsung duct (white arrow). Taken from the Imaging Service of the Metropolitan Hospital.")
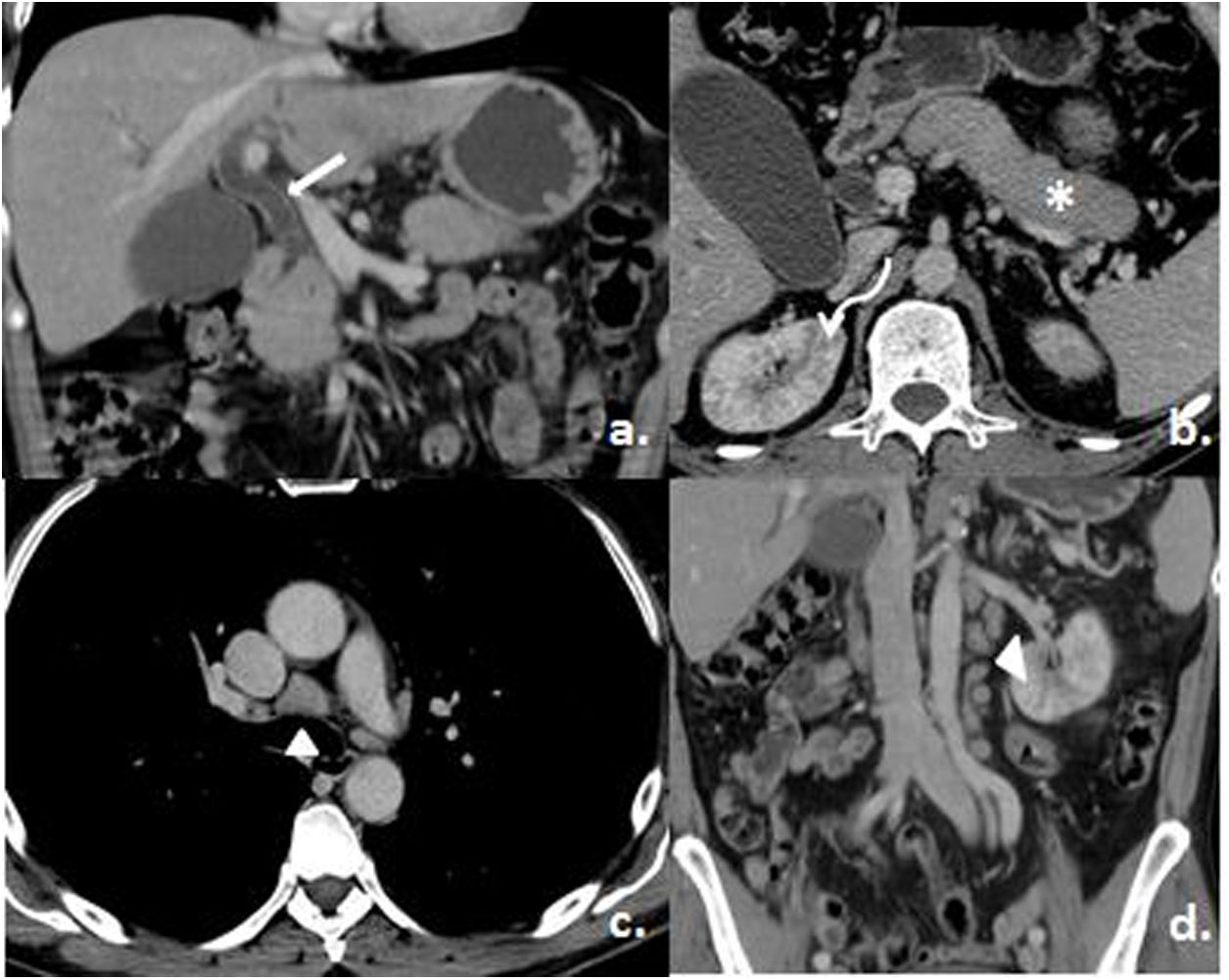
Given the significant dilatation of the biliary tract, a tomographic study was requested, which reported a dilatation of the common bile duct (choledochus) (16mm), with parietal thickening of the entire biliary tract and enhancement with the contrast. The pancreas showed a diffuse alteration in its morphology, with a more prominent area in the uncinate process and a suspicious focal lesion that could not be identified. In addition, multiple wedge-shaped areas due to lack of contrast uptake were found in the cortices of both kidneys, diffusely distributed, and multiple supraclavicular and mediastinal nodes (in the prevascular and pretracheal space, the largest at this level of approximately 2.7×1.9cm), around the celiac trunk, the hepatic hilum, and larger in the left para-aortic retroperitoneum and adjacent to the vena cava, as well as in the inguinal and iliac regions bilaterally (Fig. 2).
 Dilatation and enhancement of the common bile duct (white arrow). b) Loss of the peripheral lobulations of the pancreas (asterisk) and multiple renal perfusion defects (curved arrow). c and d) Mediastinal and retroperitoneal adenomegalies (arrowheads). Taken from the Imaging Service of the Metropolitan Hospital.")
Contrast tomography of the thorax and abdomen. a) Dilatation and enhancement of the common bile duct (white arrow). b) Loss of the peripheral lobulations of the pancreas (asterisk) and multiple renal perfusion defects (curved arrow). c and d) Mediastinal and retroperitoneal adenomegalies (arrowheads).
Taken from the Imaging Service of the Metropolitan Hospital.
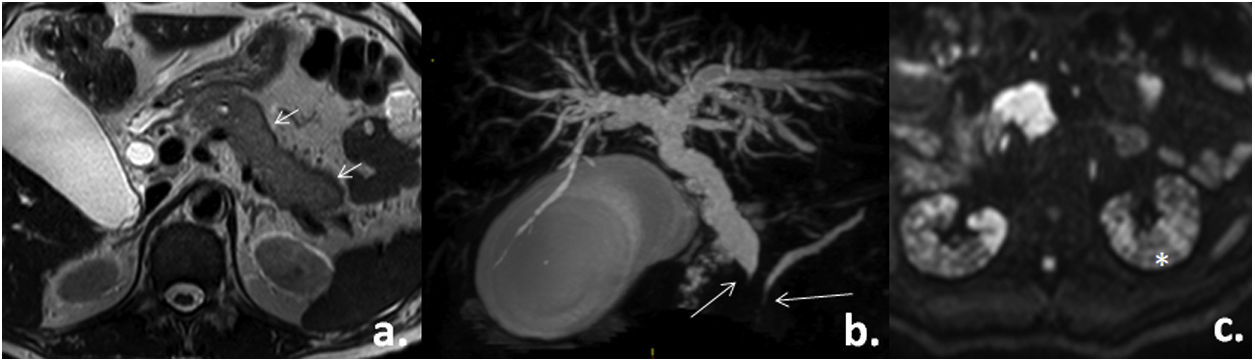
For a better visualization of the pancreas, an MRI was requested, which revealed a global loss of pancreatic architecture, with peripheral hypointensity, narrowing of the distal common bile duct with proximal dilatation (14mm caliber) and of the distal Wirsung duct. Some left para-aortic adenomegalies and patchy alteration in both kidneys were associated (Fig. 3).
 T2 axial slice: alteration of pancreatic morphology (short arrows). b) Volumetric reconstruction of the biliary tract: filiform narrowing of the common bile duct and the Wirsung duct with proximal dilatation (long arrows). C) Axial slice in diffusion: patchy alteration of the renal signal (asterisk). Taken from the Imaging Service of the Metropolitan Hospital.")
Abdominal MRI. a) T2 axial slice: alteration of pancreatic morphology (short arrows). b) Volumetric reconstruction of the biliary tract: filiform narrowing of the common bile duct and the Wirsung duct with proximal dilatation (long arrows). C) Axial slice in diffusion: patchy alteration of the renal signal (asterisk).
Taken from the Imaging Service of the Metropolitan Hospital.
When facing an adult patient with an acute clinical picture of jaundice due to cholestasis, severe thrombocytopenia, plus image findings indicative of an inflammatory process that resembles a pancreatic pseudotumor that explains the obstructive process of the biliary tract, inflammatory, autoimmune or neoplastic causes were suspected; the latter less likely since a well-structured tumor lesion without contrast uptake was not evident. Quantification of the total IgG was requested, which reported a value of 3183mg/dl (normal value: 3−201mg/dl), with an IgG4 of 887mg/dl (normal range: 3−201mg/dl) and IgE 243 IU/mL (normal <100 IU/mL); in addition to the measurement of the tumor marker Ca 19-9, which was significantly elevated (50N), and a carcinoembryonic antigen that was negative. Immunological tests were also requested, which reported hypocomplementemia with C3: 22pg/ml (normal: 0–8pg/ml) and C4: 47mg/dl (normal: 90−180mg/dl), as well as an elevation in the erythrocyte sedimentation rate by Westergreen of 59mm/1h (range 3–5) and 73mm/2h (range 7–15). Antinuclear antibodies were positive, with a 1:100 dilution; and a study of anti-DNA, anti-neutrophil cytoplasmic antibodies, anti-smooth muscle antibodies, and liver kidney microsomal type 1 antibody, was negative. On the other hand, the renal function was not affected (creatinine 1.0mg/dl); however, the patient presented a quantitative oligoalbuminuria of 153.9mg/l (range 0−20mg/l).
A study of the salivary glands was not carried out since the patient did not present any clinical symptoms; and the tomographic study did not report any finding that suggested any abnormality. Likewise, no studies for the detection of Helicobacter pylori (H. pylori) infection were requested, nor were electrophoresis of proteins and immunofixation.
Based on the clinical manifestations, the biomarkers, and the imaging studies performed, it was considered that the patient met the criteria for type I autoimmune pancreatitis associated with severe thrombocytopenia. In this context, management was started with methylprednisolone 1g intravenously daily for 3days, followed by prednisone 1mg/kg/day. The clinical evolution of the patient was satisfactory, with a progressive decrease in petechiae, ecchymosis and jaundice that correlated with the laboratory data, which reported an early decrease in cholestatic markers and a recovery from thrombocytopenia to normal from the eleventh day of treatment, as well as a decrease in IgG4 and Ca 19-9 (Table 1).
At the fourth week, it was performed an endoscopic ultrasound that showed a diffuse and somewhat nodular hyperechogenic pancreatic parenchyma. During his evolution, the patient developed diabetes secondary to the pancreatitis or to the use of corticosteroids, for which he required the establishment of insulin-based treatment.
At 2years, an ultrasound performed to the patient did not report focal lesions or dilatation of the biliary tract, with a diameter of the common bile duct of 4.2mm; after 3years the patient remains with a treatment based on prednisone (5mg/day), asymptomatic and with normal laboratory tests.
DiscussionIgG4-related disease is a rare autoimmune disorder, first recognized in 2003, characterized by lymphocytic and IgG4-positive plasma cells infiltration in almost all organs,1 including: salivary glands (submandibular, parotid and sublingual) and lacrimal glands, which together make up Mikulicz's disease, clinically characterized by pain and edema in these glands and which is sometimes confused with Sjögren's syndrome; in addition, there is involvement of the pancreas and the biliary tree, lungs, kidneys, pituitary gland, aorta, retroperitoneum, meninges, and thyroid gland (Riedel's thyroiditis).2–4
One of the most frequent presentations is autoimmune pancreatitis, clinically characterized by painless obstructive jaundice and sometimes with the onset of diabetes or symptoms of pancreatic insufficiency. 2types are described: type ii, which is the most frequent manifestation of the IgG4-related disease, in which histologically is found a lymphoplasmacytic infiltrate rich in IgG4-positive cells and fibrosis that surrounds the pancreatic lobes, its ducts and the peripancreatic adipose tissue or that of other organs; it affects more often men over 50 years of age, and type ii, which histologically shows an idiopathic ductocentric pancreatitis and, unlike type i, only affects the pancreas; occurs in younger patients, with the same proportion in the 2 genders and has few IgG4-positive cells.1,2
The association of thrombocytopenia with IgG4-related disease is very rare; its physiopathogenic mechanism is autoimmune. In some case reports, it has been described the association with the presence of antibodies on the surface of the platelet in which glycoprotein iib/iiia or ib/ix/iv is found.4 The response to corticotherapy is usually satisfactory, however, in some occasions is necessary to add other immunosuppressants.3,5
It is important to take into account that patients with an IgG4-related disease have multiorgan involvement, which in many cases can be confused with some type of malignancy, infections or other condition of immune type. This IgG4-related disease has 2 characteristics that can be confused with a gastrointestinal neoplastic disease: the first is that in the tomographic study the image that is visualized as a product of the inflammatory process sometimes resembles a tumor or mass, therefore, it is often described as a pancreatic pseudotumor, but it is not actually. And the second characteristic is that in most cases it is accompanied by elevated serum levels of Ca 19-9, which is why timely clinical differentiation between a tumor disease and an autoimmune disease is needed to avoid unnecessary treatments or surgeries.2–6
The diagnosis of this disease is based on a combination of clinical, serological, radiological and pathological findings. In this sense, the enlargement of the affected organ, masses or nodular lesions, serum levels of IgG4>1.35g/l (occurs in 55–97% of cases)6 and histopathological findings with>10 IgG4 + cells/high power fields are observed, as well as a ratio of positive cells with IgG4/IgG>40%.7 The disease can manifest itself in different ways depending on the organ involved (11 possible organs); in the recent bibliography there is a document published in the year 2019 by the American College of Rheumatology and the European League Against Rheumatism which presents the 8 inclusion criteria and the 32 of exclusion for the diagnosis of a IgG4-related disease, taking into consideration the previously mentioned findings, knowing that a certain number of patients present this disease without elevated IgG4 levels with confirmatory histopathology, which is probably due to the degree of systemic involvement.2
Among the serological exclusion criteria, thrombocytopenia and leukopenia are mentioned, because their presentation, as well described, is unusual and is generally related to other diseases, such as myelodysplastic syndromes, hematopoietic neoplasms and other autoimmune conditions such as systemic lupus erythematosus;2 however, the patient whose case is reported did not require complementary studies such as bone marrow aspiration or biopsy because after 48h of starting treatment he improved markedly until reaching normalization. For this reason, even though diagnostic criteria are mentioned, the disease can have unusual presentations such as this one, which are important to consider and not exclude at the time of the comprehensive assessment of the patient in whom this entity is suspected.
In many cases, the diagnosis of this disease has been established based solely on clinical symptoms, imaging and serology, since access to biopsy is often not possible.2 Histopathological confirmation was not performed in our patient since there was a high clinical suspicion, as well as other complementary studies that confirmed this diagnosis.
On the other hand, although it has been described the association of hypergammaglobulinemia, as well as H. pylori infection, with the development of IgG4-related disease, it is important to mention that within the diagnostic algorithm in our patient, a weakness was not having requested studies for the detection of H. pylori nor protein electrophoresis and immunofixation.
The first-line treatment recommended for a long time is with glucocorticoids, which have an anti-inflammatory effect and cause rapid improvement in symptomatology and in abnormal laboratory parameters. There is no established protocol for corticosteroid therapy: however, for the induction of remission it is recommended to start with 0.6−1mg/kg of prednisone (30−40mg/day) or some other equivalent steroid during 2–4 weeks, to then progressively reduce 5mg every 1–2 weeks, with a regular laboratory and imaging control after 4weeks, so that treatment ends in 3–6 months according to the clinical evolution. The use of methylprednisolone, at doses higher than 1g/day for 3days is also described, especially in cases in which urgent treatment is necessary and the risk of organ damage is high. In the same way, the use of 2 doses of rituximab 1g intravenously within a period of 15 days is indicated.7,8 It has been preferred to maintain this patient with the use of corticosteroid therapy, thanks to which he has remain stable and without recurrences.
It is described that 40% of the patients may suffer a relapse within the first 3years after diagnosis, either in the same affected organ or in another anatomical site, moment in which glucocorticoids can be used again in association with other immunosuppressants such as azathioprine, mycophenolate mofetil, methotrexate or mercaptopurine, thus obtaining a remission of up to 93% compared to those who only use corticosteroids, 79% at 6months. In those in whom the response to the described treatment is refractory, other immunosuppressive agents such as rituximab have been used, with a longer duration.7,8
Recently, drugs that can act according to the pathophisiological mechanism of this disease, which is more and more understood every day, have been developed or applied, taking into account that in the first phase, clearly inflammatory, cytokines such as interleukin 1 (IL-1), IL-6 and interferon gamma, among others, which will be responsible for activating lymphocytes, are released. Hence the use of drugs such as tocilizuman or anakinra. In this way, the adverse effects resulting from prolonged corticosteroid therapy can be avoided and in some cases without having the desired effect due to subtherapeutic doses.7
ConclusionIgG4-related disease is a rare disorder of autoimmune etiology, with clinical manifestations that depend on the organ involved in each patient. The most frequent presentation is type i autoimmune pancreatitis, sometimes accompanied by involvement of salivary and lacrimal glands, as well as other manifestations in different organs resulting from plasmacytic infiltration, such as those described here. Furthermore, the association with autoimmune thrombocytopenia is very rare, so it is important to identify this disease and treat it on time, to prevent the affected organs from reaching the state in which fibrosis occurs and the damage is irreversible, despite the treatment established, which can lead to organ failure and death.
From the clinical point of view, this case was difficult to diagnose, due to the low prevalence in Colombia and with no clinical findings on admission other than jaundice in association with thrombocytopenia manifested clinically with ecchymosis and petechiae. An imaging study showed a "pancreatic pseudotumor" with obstruction of the biliary tract, and due to the suspicion of an inflammatory-type disease, complementary studies were requested to confirm autoimmune pancreatitis.
The authors of this paper consider that the autoimmune spectrum of this disease is still an area under development, which requires the contribution of multidisciplinary research teams that include rheumatologists, internists, gastroenterologists, nephrologists, pneumologists, neurologists, radiologists and pathologists, and that studies of this type of disease are transcendental at the time of diagnosis and management of the patients.
Ethical ConsiderationsInformed consent was duly authorised by the patient, respecting the right to privacy. Endorsement was received from the Ethics Committee of the Metropolitan Hospital for its publication in a scientific journal.
FundingNone.
Conflict of interestThe authors do not declare any conflict of interest for the preparation of this article.
We thank Ángela León Cáceres, Master in Public Health, for her collaboration in reviewing and translating the article, as well as the Metropolitan Hospital for allowing access to the clinical history and laboratory data of the patient.
