



Macrophage activation syndrome (MAS) is a pathological systemic inflammatory reaction that is often fatal and underdiagnosed. There may be multiple organ failure that could be triggered in association with rheumatic, neoplastic or infectious diseases and/or drugs. It has been reported more in children than adults, probably as it is often associated with genetic abnormalities not described yet undescribed, genetic abnormalities. In most cases the genetic defect is not recognized in adults, or has a different etiology. The signs and symptoms of macrophage activation syndrome have been defined. Not suspecting its presence may lead to not making the diagnosis and thus, an increase in mortality. Diagnosis is a challenge, treatment has to be started early and be aggressive to reduce the high mortality rate.
ObjectivesTo describe four adult patients with five MAS episodes related to different underlying diseases, with the aim of making it familiar to the reader, to look for the syndrome and make a diagnosis.
Materials and methodsPatients evaluated in outpatients and while in the hospital.
ResultsWe present the characteristics of MAS, with the diagnostic approach and the therapeutic possibilities and their outcomes.
ConclusionsMAS is not looked for in the adult and could be fatal. It requires identification and early treatment to reduce the risk of mortality. It still needs to be studied to define the genetic defect, or other causes that may be responsible for the development of the syndrome.
El síndrome de activación macrofágica (SAM) es una reacción patológica inflamatoria sistémica, frecuentemente fatal y comúnmente no diagnosticada, que se acompaña de una falla multiorgánica y puede desencadenarse asociada a enfermedades reumáticas, neoplásicas, infecciosas o a drogas. Más descrita en niños que en adultos, probablemente en muchas ocasiones se relaciona con alteraciones genéticas aún no descritas. Sus síntomas y signos han sido definidos. El no sospecharlo conlleva a no diagnosticarlo y como consecuencia a un incremento importante del riesgo de mortalidad en el paciente; es por esto que el diagnóstico es un reto y el tratamiento debe de ser temprano y agresivo.
ObjetivosDescribir 4 pacientes adultos con 5 episodios de SAM relacionado con diferentes enfermedades reumáticas, con el interés de familiarizar al lector con la búsqueda del síndrome y de realizar su diagnóstico.
Materiales y métodosEstudio descriptivo de pacientes adultos evaluados en la consulta y hospitalizados.
ResultadoPresentamos las características de los pacientes con SAM, el enfoque diagnóstico, las posibilidades terapéuticas y la evolución.
ConclusionesEl SAM es una enfermedad no buscada en el adulto que puede ser fatal, requiere ser identificada y tratada tempranamente para disminuir el riesgo de mortalidad. Aún requiere ser estudiada para definir defectos genéticos u otras etiologías que puedan ser responsables de este síndrome.
Familial lymphocytosis is a pathological immune activation with signs and symptoms of severe inflammation, which was recognized in 1952 as a childhood disease. In 1977, the macrophage activation syndrome (MAS) is reported in isolated cases presenting hepatic insufficiency and consumptive coagulopathy,1 subsequently, MAS is described as a problem related with juvenile rheumatoid arthritis.2 In 1993, it is described in detail and specifically in 24 children.3 Today it is quite well characterized becoming a clinical entity associated with or triggered by drugs, infections, cancer or rheumatic diseases, or existing as an isolated disease without a trigger.4 MAS has been described more frequently in children than in adults, but there has been an increase in reported adult patients.5
MAS is a clinical picture similar to the hemophagocytic syndromes, in which a perpetuation of the inflammatory response occurs due to a primary deficiency or a defect in the apoptosis of activated macrophages. This primary defect has been defined in several cases; in others, its etiology is still to be defined. MAS occurs when there is a poorly controlled macrophage function, a reduced cytotoxic cell activity and a decrease in the toxic T lymphocytes and natural killer cells. Some cytokines and amplifications of the inflammatory response have been described.6 The apoptosis of activated macrophages is achieved by the pathway of the perforin pore-granzyme B serine protease. The reduction in the number or in the function of NK cells or a genetic abnormality of the granzyme-perforin pathway leads to a MAS.
In juvenile idiopathic arthritis (JIA), lupus erythematosus and other rheumatic diseases there is an increase in the incidence of MAS.7,8 The MAS, is often confused with the inflammation in sepsis or with the exacerbation of the underlying disease.9 The syndrome has been described by two different groups: the Histiocytosis Society and the pediatric rheumatologists. The guidelines for the diagnosis have been published by the two trends and are in the process of definition for systemic lupus erythematosus (SLE) and systemic JIA.10,11 Recently, the 2 groups came together and defined the nomenclature, as well as the diagnostic criteria.12–14 The diagnosis is based on the presence of clinical and laboratory criteria, already established by the Society of Lymphohistiocytic Hemophagocytosis.15 Ravelli et al., developed the criteria for MAS associated with JIA.11
Description: the MAS has an acute onset and its clinical manifestations are: high and persistent fever (94%), neuropsychiatric changes, splenomegaly (59%), hepatomegaly (88%), lymphadenomegaly (48%), skin erythema (65%), decrease in the number of cells of 2 or 3 cell lines at the peripheral blood level, anemia (82%), thrombocytopenia (88%) (>150,000 platelets); leukopenia (56%), elevated liver enzymes (transaminases) (46%), low fibrinogen due to consumption (100%), hypertriglyceridemia (80%), prolonged prothrombin and thrombin time and very high ferritin levels (100%) (in parentheses are the percentages of these characteristics that are described in the criteria for MAS associated with JIA by Ravelli.)11 In a recent article, hemophagocytic lymphocytosis is recognized as a disorder with increasing diagnosis in the adult.16
It has been suggested recently that the serum levels (determined by ELISA) of the specific receptor waste collector (scavenger) of macrophages, the soluble sCD163, are specific markers for macrophages in patients who have an inadequate macrophage activation, being very elevated and serving their determination for the follow-up of the activation and the disease activity.17,18 Likewise, the determination of levels of the soluble receptor for the interleukin-2 receptor (sIL-2R), has been used as a marker.18
Concerning imaging, changes in various systems and structures related to the excessive inflammatory response in the affected organs have been described.19 The chest X-ray shows an interstitial commitment that is compatible with local inflammation or may be related to interstitial hemorrhage. In the resonance of the skull, images of high density of with lack of tissue can be seen in cases with involvement of the brain; these findings have been described in patients with acquired immunodeficiency syndrome, and we have seen them in our patients.
In spite of being established the criteria, they still require to be validated for diseases other than systemic JIA. There are situations that make difficult the diagnosis, among others, at the beginning of the disease it may or may not exist phagocytic activity in the bone marrow, the liver or the spleen (the last two are not considered for biopsies). Hemophagocytosis may not be detected in these early stages and it can be non-specific in the case of a blood transfusion. It may be confused with activity of the underlying disease or with sepsis. The elevation in triglycerides, the determination of natural killer cells and the cytotoxicity of T-cells, or the CD163 or the interleukin-2 receptors, are not procedures that are carried out on routine basis in the laboratories. Ferritin is not a routine test, and is not performed urgently and may take several days before obtaining the results. That makes it important to consider the diagnosis of MAS, as can be seen in the following cases.
The mortality of MAS is high, from 32% to 80% depending on the reviewed series. The lack of knowledge of the existence of this syndrome, the delay in the diagnosis and the delayed onset of therapeutic measures may be responsible for this high mortality. Silva presented a small number of patients in whom the high mortality coincides with the delay in the diagnosis, hence the need for an early diagnosis.20
The Epstein Barr virus is one of the most commonly described and even it has been held responsible as a trigger agent. The National Institute of Health has a website where the cases associated with infections are cited. On the other hand, the underlying diseases, cancer, leukemia, lymphoproliferative disease, SLE, juvenile dermatomyositis, JIA and rheumatoid arthritis (RA), are associated with MAS. It can be triggered by whichever medication, aspirin and other non-steroidal anti-inflammatory drugs, the second injection of gold salts, sulfasalazine, methotrexate21 and anticonvulsants. It is noteworthy that the MAS is associated with the biologicals anti-tumor necrosis factor, infliximab, etanercept o anti-interleukin-1, anakinra, since these are the cytokines that are triggered in the processes of inflammation and the latter is used today in many centers as the therapy of choice for the syndrome. Theoretically, the anti-TNF could be considered as a therapy by the same pathophysiological description of the process, but great discussion on this regard has aroused due to the paradoxical response described with these treatments; some patients had the induction of MAS with anti-TNF therapy.22,23 On the other hand, a dramatic response to etanercept occurred in a 7-year-old boy with JIA.24 In our series is described the EG patient, who develops the syndrome after starting treatment with anti-TNF as a single drug.25
Treatment is directed primarily to the elimination of the cause that induced the syndrome. Then starts the treatment with pulses of methylprednisolone at a dose of 30mg per kg of body weight or up to one gram per day for 3–5 days, depending on the severity of the MAS and on the response to the initial doses. Oral prednisone can be used at 1 or 2mg per kg of body weight per day, in single or divided doses for more effect, depending on the severity of the disease. The pulse can be repeated in case of insufficient response. Cyclosporine has been used successfully due to its stabilizing effect on the macrophage membrane or its function on the helper lymphocytes.26,27 Immunoglobulins and etoposide have also been used (the latter mostly by the Hemophagocytosis Society). Anakinra, which is a human interleukin-1 receptor antagonist (IL-1Ra) is widely used today. Tocilizumab, a recombinant humanized monoclonal antibody against the anti-interleukin-6 receptor (IL-6), is used in the treatment of some patients who do not tolerate interleukin-1 or for whom it is not affordable (Table 1)
Manifestations and demographic data of MAS in the different patients.
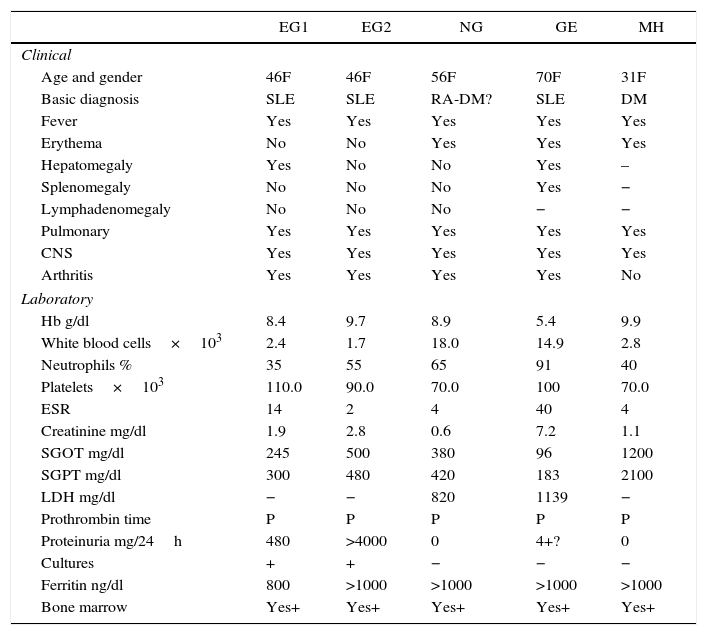
| EG1 | EG2 | NG | GE | MH | |
|---|---|---|---|---|---|
| Clinical | |||||
| Age and gender | 46F | 46F | 56F | 70F | 31F |
| Basic diagnosis | SLE | SLE | RA-DM? | SLE | DM |
| Fever | Yes | Yes | Yes | Yes | Yes |
| Erythema | No | No | Yes | Yes | Yes |
| Hepatomegaly | Yes | No | No | Yes | – |
| Splenomegaly | No | No | No | Yes | − |
| Lymphadenomegaly | No | No | No | − | − |
| Pulmonary | Yes | Yes | Yes | Yes | Yes |
| CNS | Yes | Yes | Yes | Yes | Yes |
| Arthritis | Yes | Yes | Yes | Yes | No |
| Laboratory | |||||
| Hb g/dl | 8.4 | 9.7 | 8.9 | 5.4 | 9.9 |
| White blood cells×103 | 2.4 | 1.7 | 18.0 | 14.9 | 2.8 |
| Neutrophils % | 35 | 55 | 65 | 91 | 40 |
| Platelets×103 | 110.0 | 90.0 | 70.0 | 100 | 70.0 |
| ESR | 14 | 2 | 4 | 40 | 4 |
| Creatinine mg/dl | 1.9 | 2.8 | 0.6 | 7.2 | 1.1 |
| SGOT mg/dl | 245 | 500 | 380 | 96 | 1200 |
| SGPT mg/dl | 300 | 480 | 420 | 183 | 2100 |
| LDH mg/dl | − | − | 820 | 1139 | − |
| Prothrombin time | P | P | P | P | P |
| Proteinuria mg/24h | 480 | >4000 | 0 | 4+? | 0 |
| Cultures | + | + | − | − | − |
| Ferritin ng/dl | 800 | >1000 | >1000 | >1000 | >1000 |
| Bone marrow | Yes+ | Yes+ | Yes+ | Yes+ | Yes+ |
NA: not obtained; No: absent; Yes: present; P: prolonged; +: positive; −: negative.
Age: 46 years. Gender: female. Consults for: disorientation, left hemiparesis, polyarthritis and fever. At physical examination is conscious, confused and disoriented. BP: 150/110mmHg, heart rate: 87×minute, weight: 40.300kg, temperature: 39.8°C, severe livedo reticularis, alopecia, and two painless, remittent, non-adherent cervical lymphadenopathies. Rhythmic muffled heart sounds without murmurs. Vesicular murmur present and symmetrical with bilateral crackles. Soft, depressible abdomen; painful palpable hepatomegaly. Nonpalpable spleen. Arthritis in the ankle, the knee and the left wrist. Pitting edema in the left ankle. Muscle strength in the left lower limb 3/5, left upper limb 4/5. Osteotendinous reflexes: 3/4 left lower limb; all other reflexes 2/4. No evidence of varices.
History previous to hospitalization: eight months before she presented asthenia, arthritis of the hands, knees and ankles, weight loss of 30kg, dry cough, oppressive retrosternal pain that worsened with decubitus, dyspnea of medium efforts, recurrent fever of 39 degrees, behavioral disorders and pancytopenia, diagnosed as lupus and dengue hemorrhagic fever. Laboratories: Hb: 8.4g/dl, WBC: 2.4×103; neutrophils: 35%; platelets: 110,000. ESR: 14mm, SGOT: 245mg/dl, SGPT: 300mg/dl. Creatinine: 1.9mg/dl. Proteinuria: 480mg/dl. Blood and urine cultures with Salmonella species. Chest X-rays: reticulonodular infiltrate. Bronchoscopy: alveolar-bronchial hemorrhage. Not measurable prolongation of prothrombin time and partial thromboplastin time and ferritin levels of 800ng/ml (normal: 233ng/ml, Abbot axsym systems). The bone marrow aspirate shows the presence of macrophages phagocytizing formed elements.
Diagnosis at hospitalization: SLE, sepsis due to Salmonella, nephrotic syndrome, cerebral vasculitis, hemorrhagic diathesis, pulmonary hemorrhage and MAS, and treatment with cyclosporine, antibiotics and general measures is started, with improvement.
Two weeks later the patient is well and stable, not presenting symptoms, coherent, optimistic, cheerful, alert, very recovered, with some weakness and no alterations on physical examination, without arthritis, with almost total improvement of the left hemiparesis and her laboratory tests results show: hemoglobin 8.70g/dl, WBC 3400 segmented 65, lymphocytes 33, platelets 300,000, creatinine 1.6mg/dl. Transaminases are normal. An immunological profile shows positive antinuclear antibody, positive anti-DNA, and decreased complement values.
Prior to the second hospitalization, one month after the first one: the patient stops taking all her medicines and after 7 days presents a seizure and fever. Her laboratory values are: Hb 9.7g/dl, Cr 2.8mg/dl. White blood cells 1.7×103; neutrophils 55%, platelets 90×103, sedimentation 2mm, SGOT 500mg/dl, SGPT 480mg/dl; albumin 1.8mg/dl, proteinuria >4g/24h. Ferritin >1000ng/ml. Is hospitalized with a multiple organ failure: pulmonary hemorrhage, hemorrhagic diathesis, severe renal failure, nephrotic syndrome and hypertension, left hemiparesis, cognitive and behavioral disorders. Second hospitalization diagnosis: MAS, systemic vasculitis, SLE complicated with nephropathy, arterial hypertension.
Second hospitalization treatment: steroids, immunoglobulins, cyclosporine and corrective measures for her multiple organ failure, tracheostomy is performed and she receives positive-pressure mechanical ventilation and requires dialysis. She has complication with sepsis caused by Staphylococcus aureus and Escherichia coli; the patient eventually does not respond to treatment and dies.
Case 2 NGAge: 56 years. Gender: female. Consults for: confusion after a seizure.
Previous history: six months earlier is diagnosed with RA, receives treatment with methotrexate for a month and due to elevated transaminases is changed to sulfasalazine, hydroxychloroquine and prednisone. Four months earlier, she discontinues everything and receives treatment with unknown drugs, of natural origin, so-called: “systemic medicine”, with the development of symptoms such as sleeping difficulty, pruritus and bradypsychia, difficulty in walking, difficulty to sit up, violaceous redness around the eyes and the ear with erythema all over the body. She has no history of allergy or other disease.
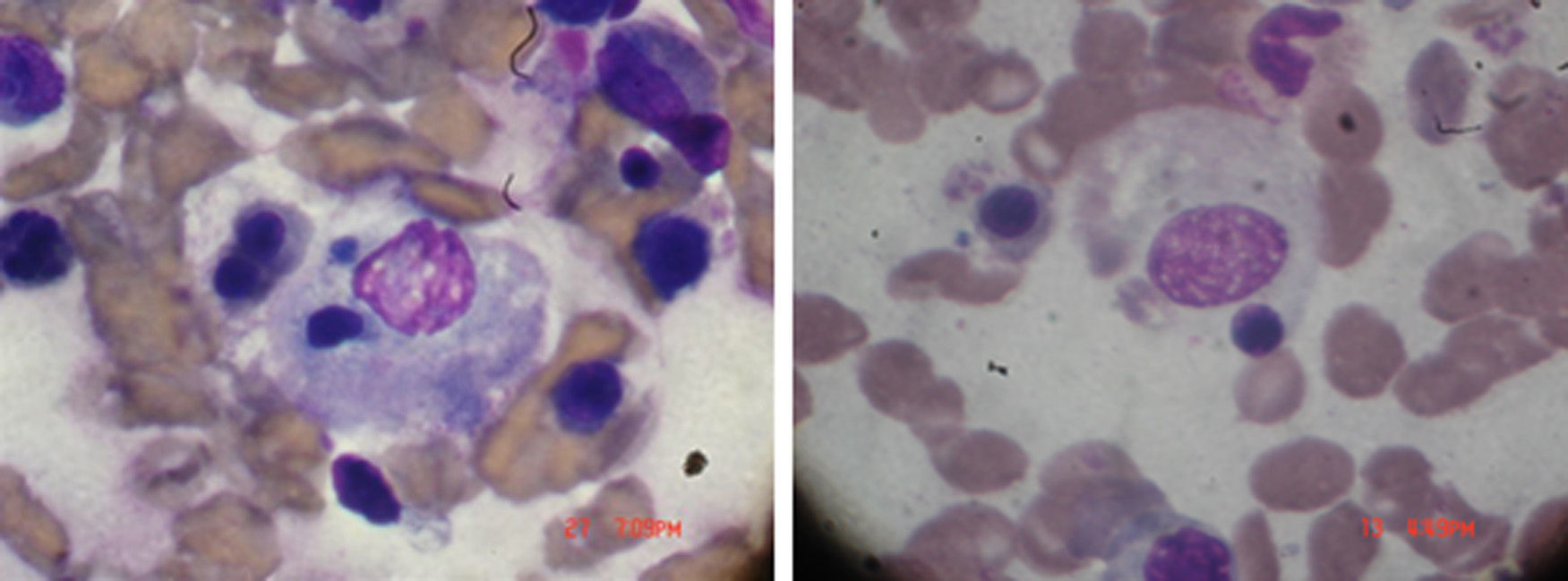
Physical examination on admission: temperature: 38.5°C. Weight: 92kg. BP: 110/60. Periorbital heliotrope edema, desquamative erythema in the pinna and erythema on the back of the hand and in the anterior and posterior region of the chest, good expansion of the thorax with crackles in the left base. No cardiomegaly, regular and rhythmic heart sounds with tachycardia. Soft, depressible abdomen without visceromegaly or signs of neurological focalization; articular exam with synovitis of proximal interphalangeal and metacarpophalangeal joints. Laboratories: hemoglobin: 8.9g/dl, with white blood cells 18×103, 65% neutrophils, platelets 70×103, SGOT 380mg/dl and SGPT 420mg/dl, with a prolongation of PTT, erythrocyte sedimentation rate 4mm, high triglycerides, 560mg/dl, LDH 820mg/dl creatinine 0.7mg/dl. Creatine-Phosphokinase 1080mg/dl. Ferritin >1000ng/ml. In bone marrow presence of macrophages phagocytizing blood elements, as shown in Fig. 1. Diagnosis: RA, with findings of dermatomyositis and MAS. Treatment and volution: methylprednisolone one gram for 4 days. Cyclosporine 4mg/kg intravenously is added and then maintenance doses of both drugs, with full improvement of the clinical picture. Cyclosporine was maintained, since whenever we tried to discontinue the medication, the patient showed signs of exacerbation of the MAS. The patient remained stable without crisis of the disease with the cyclosporine-based treatment.
Case 3 GE
Age: 70 years. Gender: female. Reason for consultation: dyspnea, changes in the skin (fragile, “pressed” with loss of dermal annexes in the forearm, hands, ankles and feet), Raynaud's phenomenon and arthralgia, 7 months prior to lung biopsy that shows interstitial fibrosis. No history of hypertension, renal disease, dyspepsia, difficulty swallowing, or calcifications. Allergy to non-steroidal drugs. Physical examination: BP: 110/70mmHg. Pulse 80×min, Respiratory rate: 20×min. Reduced chest expansion with crackles in both bases. She has not cardiomegaly and the heart sounds are regular and rhythmic without murmurs or gallop. Laboratories: they are normal with the presence of an anti sl-70. A diagnosis of scleroderma is made and therapy with prednisone 7.5mg per day is started, to control arthralgia and avoid the use of non-steroidal anti-inflammatory drugs, and taking into consideration that steroids may precipitate renal crises in these patients, when used at high doses. With no major changes or new symptoms, shows improvement of the arthralgia 2 weeks after the lung biopsy. Treatment: methotrexate (it could not be administered) and etanercept.
Evolution post-treatment with etanercept: after receiving the first dose of etanercept, she develops pruritus on arms and shoulders for a period of 8h and 50h later is evaluated for abdominal pain and vomiting, somnolence, anorexia, dehydration and weakness. Respiratory arrest, develops anuria and is admitted in the intensive therapy unit, due to multisystem failure. Physical exam: blood pressure 90/60mmHg, heart rate of 120×min, respiratory rate of 40×min, afebrile. No new lesions in the skin or arthritis; bilateral crackles in both lung fields, with an increase of the first sound and a II/IV systolic murmur without gallop. The abdomen is soft with hepatomegaly. No enlargement of the spleen. Without abnormal neurological signs. Laboratories: hemoglobin: 5.49g/dl, hematocrit: 21.9%, red blood cells: 2.19×103, white blood cells: 14.9×103, neutrophils: 91%, lymphocytes 5%, platelets: 100,000/μL, sedimentation rate: 40mm, glycemia: 109mg/dl; nitrogen: 73mg/dl; creatinine: 7.2mg/dl; calcium: 8.8mg/dl; phosphorus: 9.8mg/dl; cholesterol: 169mg/dl; triglycerides: 210mg/dl. Bilirubin: 1.78mg/dl; SGOT: 96U/l; SGPT: 183U/l; alkaline phosphatase: 122U/U; LDH: 1139mg/dl; creatine kinase: 294U/l; total protein: 5.9g/dl; albumin: 2.9g/dl; globulins: 3g/dl; sodium: 132mmol/l; potassium: 5.7mmol/l; chloride 97mmol/l; urinalysis: proteins 4+; hemoglobin 4+. Chest X-rays with bilateral increased interstitial pattern. The blood cultures were negative. The bone marrow aspirate shows multiple macrophages phagocytizing cellular elements. Admission diagnosis: multisystem failure, MAS, pulmonary hemorrhage, mixed metabolic and respiratory acidosis.
Treatment: cyclosporine and methylprednisolone.
Evolution: negative cultures. Ferritin: 852ng/ml. Anuria, dialyzed 48h after hospitalization. Bleeding diathesis, pulmonary hemorrhage, and is intubated for mechanical ventilation with positive pressure. Platelets 60,000 and are not modified. Melena. Not measurable prothrombin time. Ferritin increases to 1800ng/ml. Intravenous immunoglobulin is added to treatment without clinical response. The pulmonary hemorrhage is exacerbated, remains anuric and dies 12 days after her admission.
Case 4 MHAge: 31 years. Gender: female. Reason for consultation: inability to sit up and weakness when raising the arms above the shoulders. Confused and somewhat disoriented in time, as well as in space, and behaves very introverted and depressed.
Previous diagnosis: dermatomyositis, post-surgery of thyroid cancer on replacement therapy.
Physical examination: BP: 120/80mmHg, pulse: 68, weight: 78kg. Temperature: 37.9°C. Gottron's papules, heliotrope periocular erythema and erythematous lesions with severe muscle pain in abdominal wall and thighs. No evidence of arthritis, the neurologic exam is normal, muscle strength preserved in feet, hands, forearms and legs.
Laboratory: hemoglobin: 11.8g/dl. White blood cells: 6×103, 65% neutrophils, platelets 140×103, SGOT 3800mg/dl and SGPT 4200mg/dl, erythrocyte sedimentation rate 30mm, triglycerides 200mg/dl, LDH 820mg/dl. Creatine phosphokinase 2800mg/dl. Treatment: methylprednisolone and methotrexate. She has a post-treatment clinical picture that is interpreted as a reaction similar to dengue viral syndrome. Laboratories: hemoglobin 9.9g/dl, with leukocytosis, 2.8×103 WBC, 40% neutrophils, platelets 70×103, SGOT 1200mg/dl and SGPT 2100mg/dl, sodium 128mg/dl, erythrocyte sedimentation rate 4mm, triglycerides 430mg/dl, creatinine 1.1mg/dl. Normal urine test. Ferritin >2000ng/dl. Bone marrow: hemophagocytosis.
Diagnosis: MAS
Treatment: Methotrexate is replaced with cyclosporine orally 6mg per kg of body weight (200mg 2 times daily), however, her response is slow and begins to have changes related to steroids and cyclosporine. It is decided to add Mabterra, a murine/human anti CD-20 monoclonal antibody, which has been used in some cases as the last resort for the clinical picture in 21 days, cyclosporine was maintained for several years, without recurrence of the syndrome.
Discussion4 cases of patients with rheumatic diseases, with 5 episodes of systemic inflammatory reaction and with diagnostic elements for MAS are presented, all receive treatment with methylprednisolone and cyclosporine and 2 of them deceased.
The first case EG, with SLE, was undertreated for her illness, and presents the MAS with no apparent trigger, responding properly to therapy and relapsing upon discontinuation of treatment, the patient dies. In the second case NG, it seems to exist a correlation between the use of the “systemic medicine” and the onset or trigger of the syndrome. The treatment with cyclosporine for the control of her disease is made continuously and permanently, after the improvement of the acute condition. The case 3 GE, becomes ill after etanercept. The triggering of the syndrome is attributed to etanercept because of the cause-effect correlation that occurred and because it was the only drug in use.25 In the fourth case MH, the syndrome is triggered by the methotrexate, as already described in the literature.
The MAS is underdiagnosed. Physicians are not yet familiar with this diagnosis, especially in adults. The diagnosis should be considered in those patients with multi-organ failure, with symptoms of inflammatory origin with an autoimmune or malignant disease, especially if there is evidence of reactivation of the underlying disease with or without association with a viral, bacterial or fungal disease, or with the use of a medication. It is important to evaluate the patient properly and request the described exams, in order to make the diagnosis and administer the adequate therapy.
Having made the diagnosis of MAS timely allows to administer the proper treatment, being able to decrease the morbidity and mortality from the syndrome. Some adult patients may still have the manifestation of a genetic problem. It is very likely that defining the defect that triggers the systemic inflammatory reaction would be crucial in the therapeutic decision of the future and for the differentiation between the diverse inflammatory conditions. Although it is true that the treatment in general is more or less similar, the decision to use steroids, cyclosporine, etoposide, immunoglobulin or bone marrow transplant is crucial for the treatment of MAS.
There are many things we must learn about MAS, its etiology and treatment.
Ethical responsibilitiesProtection of people and animalsThe authors declare that no experiments were performed on human beings or animals for this research.
Data confidentialityThe authors declare that they have followed the protocols of their workplace on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the informed consent from patients and/or subjects referred in the article. This document is held in the possession of the corresponding author.
Conflict of interestThe authors declare that they have no conflict of interest.
Please cite this article as: Sterba G, Sterba Y, Iglesias G. A. Síndrome de activación macrofágica en adultos con enfermedad reumática. Rev Colomb Reumatol. 2016;23:137–143.
