



La osteoporosis es una patología que afecta el sistema esquelético y se caracteriza por una baja densidad mineral ósea y un deterioro estructural del tejido óseo. Esta enfermedad está asociada con un alto riesgo de fracturas que comprometen seriamente la calidad de vida. La incidencia de osteoporosis es actualmente mayor debido al aumento de la esperanza de vida en el mundo.
Numerosas investigaciones con respecto a nuevas estrategias de tratamiento han sido desarrolladas y tienen como objetivo inhibir la resorción ósea excesiva o aumentar la formación de hueso. Entre los tratamientos más prometedores están denosumab, un anticuerpo monoclonal que ejerce su acción contra el activador del receptor del ligando NF-kappa B, una citoquina clave de los osteoclastos; odanacatib, un inhibidor específico de la proteasa catepsina K de los osteoclastos; y anticuerpos monoclonales contra las proteínas antiesclerostina, la glucógeno sintasa quinasa-3b y dickkopf-1, dos inhibidores endógenos de la formación de hueso. En esta revisión se analizan y se revisan los conceptos actuales de las nuevas terapias.
Osteoporosis is a skeletal system pathology characterised by low bone mineral density and tissue structural deterioration. This condition is associated with high fracture risk that severely compromises quality of life. Osteoporosis incidence is becoming more significant with increasing lifespan worldwide.
Novel treatment strategies have been developed that aim to inhibit excessive bone resorption and/or increase bone formation. The most promising novel treatments include: denosumab, a monoclonal antibody for receptor activator of NF-κB ligand, a key osteoclast cytokine; odanacatib, a specific inhibitor of the osteoclast protease cathepsin K; and antibodies against the proteins sclerostin, GKS-3b and dickkopf-1, two endogenous inhibitors of bone formation. This review discusses these new therapies.
Se realizó una revisión de la literatura, encontrando artículos de relevancia sobre nuevas terapias en osteoporosis para, posteriormente, realizar una descripción amplia del tema. La revisión se llevó a cabo en las bases de datos de MEDLINE y EMBASE. La estrategia de búsqueda no tuvo límites de fecha y se realizó mediante términos MeSH de la siguiente manera: «osteoporosis AND antiresorptive therapies», «osteoporosis AND anabolic therapies», «osteoporosis AND denosumab», «osteoporosis AND cathepsin k inhibitors», «osteoporosis AND odanacatib», «osteoporosis AND relacatib», «osteoporosis AND balicatib», «osteoporosis AND Src kinase inhibitors», «osteoporosis AND saracatinib», «osteoporosis AND GLP-2 analogs», «osteoporosis AND dickkopf-1 inhibitor», «osteoporosis AND GSK-3b inhibitors», «osteoporosis AND anti-sclerostin antibodies», «osteoporosis AND blosozumab», «osteoporosis AND romosozumab».>
Además, se incluyó en la estrategia de búsqueda «new therapies in osteoporosis».
Se establecieron como criterios de inclusión artículos con las siguientes características: que se tratara de una revisión sistemática o artículo de interés para los investigadores y que cubriera el tema de nuevas terapias en osteoporosis, se excluyeron artículos con un idioma diferente al inglés o al español.
Después de ser seleccionados los artículos definitivos, se procedió a describir sus hallazgos más importantes de forma cualitativa.
IntroducciónLa comprensión de los procesos de control intracelular que regulan la remodelación ósea es esencial para la planificación de la terapia contra la osteoporosis1.
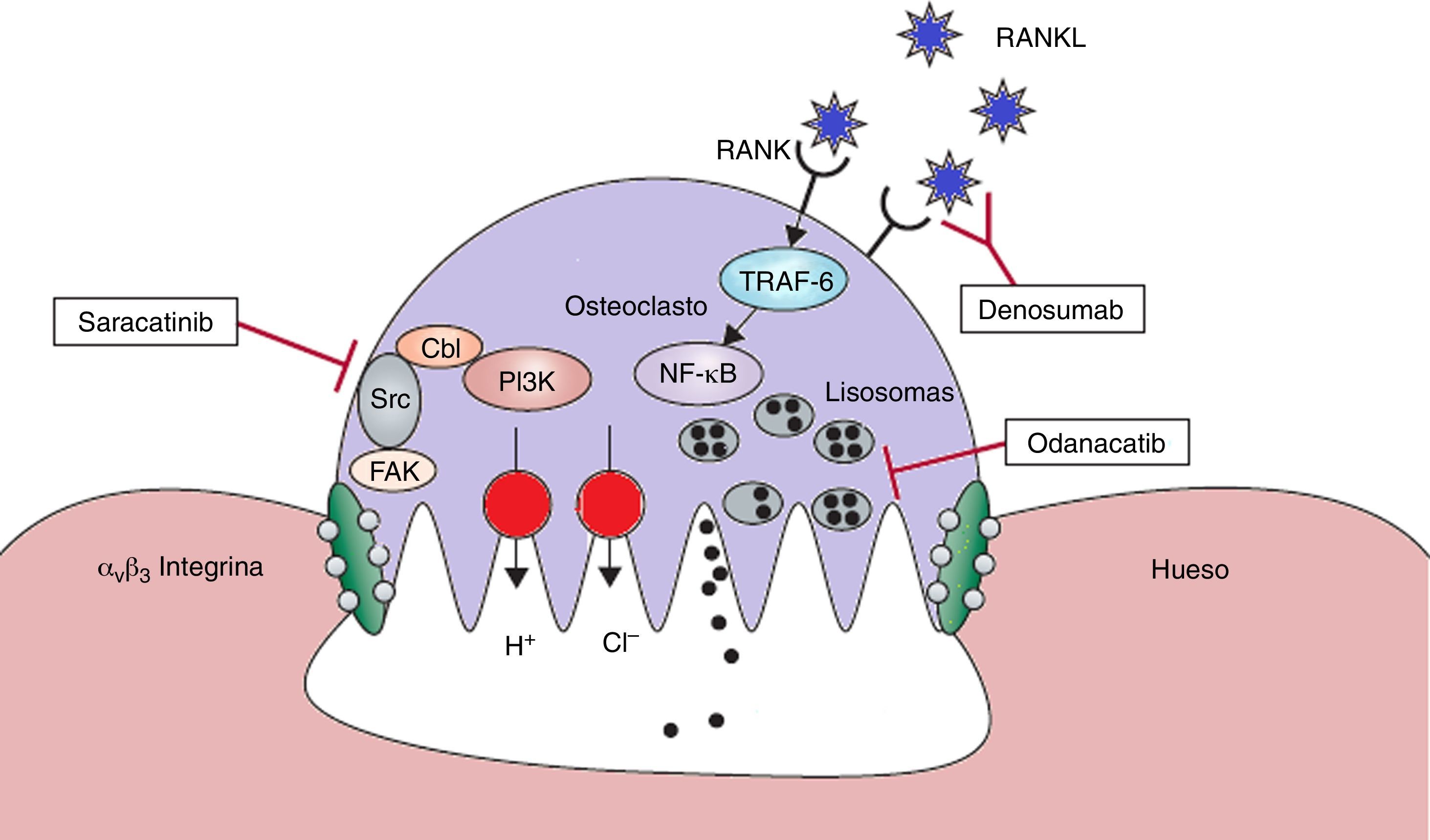
La remodelación ósea consiste en un acoplamiento estricto en la reabsorción y la formación de hueso, que continúa durante toda la vida, este proceso es necesario para la extracción de hueso dañado y obsoleto, además de mantener la estructura ósea normal. El proceso comienza con la resorción de un volumen de hueso por los osteoclastos, seguida de nueva formación de hueso por los osteoblastos2–4. Las nuevas terapias se desarrollan sobre mecanismos de acción que son diferentes a las de los medicamentos ya existentes. Algunos de estos podrían ofrecer una inhibición de la resorción sin reducir la formación de hueso (fig. 1).

Blancos terapéuticos en el osteoclasto. Con ayuda de avβ3 integrina, los osteoclastos se adhieren a la superficie del hueso y forman zonas de sellado. Se produce un microentorno altamente ácido que es esencial para la actividad catalítica de las enzimas tales como la catepsina K. El odanacatib inhibe a catepsina K, una proteasa lisosomal que degrada colágeno. Src quinasa tiene un papel crucial en la actividad de los osteclastos y puede ser inhibida por el saracatinib. RANKL actúa como regulador esencial en la diferenciación y actividad de los osteoclastos. El denosomab, anticuerpo monoclonal totalmente humano, impide que RANKL se una a su receptor.
FAK: Kinasa de adhesión focal; NF·kB: factor nuclear kappa B; P13K: fosfatidilinositol 3-quinasa; RANK: receptor activador del NF-kB; RANKL: ligando RANK; TRAF-6: receptor asociado al factor de necrosis tumoral alfa -6; Src: Kinasa que regula el crecimiento celular.
Adaptada y modificada de Rachner TD, Khosla D, Hofbauer LC. Osteoporosis: Now and the future. Lancet. 2011; 377: 1276-87.
Significa que ahora están disponibles medicamentos que permiten promover la formación de hueso nuevo, estas terapias han sido denominadas terapias anabólicas y se han generado gracias a los estudios y descubrimientos recientes en biología ósea, los cuales son prometedores frente a la identificación de nuevas dianas moleculares de este tipo de terapia5–7.
Terapia antirresorción óseaDenosumabEs un anticuerpo monoclonal totalmente humano, que tiene una alta afinidad al RANKL, molécula conocida por su destacado papel en la osteoclastogénesis8,9.
Las propiedades farmacocinéticas son superiores, comparado con otros medicamentos, lo que se traduce en un intervalo de dosificación más largo. Es el más avanzado de todos los compuestos en investigación y ha sido aprobado en Europa para el tratamiento de la osteoporosis y en Estados Unidos para el manejo de la osteoporosis y las metástasis óseas10.
Suprime, de una manera dependiente de la dosis, el telopéptido N-terminal del colágeno tipo 1 urinario (NTXu), molécula que también es usada como marcador bioquímico de la resorción ósea, hasta en el 81% en las mujeres posmenopáusicas11,12.
El denosumab en mujeres aumenta la densidad mineral ósea (DMO), en la columna lumbar, entre 3,0-6,7%, comparado con la pérdida de 0,8% en el grupo placebo. En el total de la cadera, la DMO aumentó en 1,9-3,6% en el grupo de denosumab, pero disminuyó en 0,6% en el grupo placebo13,14.
En un estudio aleatorizado de fase 3, placebo controlado (FREEDOM) denosumab (60mg cada 6 meses) se evaluó la reducción de fractura en 7.868 mujeres con osteoporosis posmenopáusica, de las cuales el 24% tenían fracturas vertebrales preexistentes. Después de 3 años, denosumab había reducido el riesgo de nueva fractura vertebral en 68%, las fracturas de cadera en 40% y no vertebrales en 20%. Es de destacar que el riesgo de eventos de enfermedades cardiovasculares, cáncer e infecciones no difiere entre los dos grupos. Sin embargo, la incidencia del eczema fue 3,0 vs. 1,7% y celulitis, incluyendo la erisipela, fue 0,3 frente a <0,1% siendo significativamente mayor en las mujeres que recibieron denosumab. La evaluación completa del estado inmunitario de los pacientes que recibieron denosumab, durante 12 meses, no mostró cambios relevantes en las células blanco, células T, células B o células natural killer (NK)15.
La extensión del estudio FREEDOM está evaluando la eficacia a largo plazo y la seguridad de denosumab para un máximo de 10 años. En un estudio abierto de 4.550 mujeres se presentan los resultados de los primeros 3 años de la extensión, en representación de hasta 6 años de exposición. El tratamiento con denosumab durante 6 años se mantuvo bien tolerado y continuó aumentando la DMO. La incidencia de fracturas se mantuvo baja, sin aumento en la incidencia de eventos adversos16.
La monoterapia o terapia combinada de teriparatide y denosumab, fue evaluada en el estudio DATA. La terapia combinada aumentó la DMO en la columna y cuello femoral más que cualquier agente; podría ser útil para el tratamiento de pacientes con alto riesgo de fractura. El diseño inicial del estudio no tenía la finalidad de detectar los efectos sobre el riesgo de fractura, siendo otra potencial limitación del estudio su diseño abierto. Por último, aunque el perfil de seguridad de la terapia combinada parece ser similar a los perfiles de los grupos de terapia individual, este estudio podría no evaluar adecuadamente la seguridad a largo plazo de cualquiera de las intervenciones. Para ello sería necesario una cantidad mucho mayor de pacientes en un ensayo clínico17.
Después de dos años de extensión del estudio DATA, 94 mujeres presentaron un aumento de la DMO más que en la monoterapia. La combinación de estos agentes puede llegar a ser una opción interesante de tratamiento en pacientes con alto riesgo de fractura18.
Los resultados demuestran que el denosumab, a diferencia de los bifosfonatos, se asocia con supresión máxima de la resorción ósea, incluso cuando se administra con teriparatide17,18.
Dos estudios aleatorios, placebo controlado se han embarcado en el uso de denosumab en mujeres que reciben inhibidores de aromatasa para cáncer de mama y hombres en terapia de ablación androgénica para cáncer de próstata.
En las mujeres con inhibidores de la aromatasa para el cáncer de mama no metastásico, denosumab (60mg cada 6 meses, durante 12 meses) aumentó la DMO en la columna lumbar en 5,5% en comparación con el placebo, este estudio no fue diseñado para evaluar la reducción de fracturas19.
En el estudio HALT, se evaluaron hombres bajo terapia de privación de andrógenos para cáncer de próstata, denosumab (60mg cada 6 meses, durante 24 meses) también aumentó la DMO en la columna lumbar en 6,7%, en el total de cadera en 4,8% y en el tercio distal del radio en 5,5%, en comparación con el placebo. Se redujo la incidencia de nuevas fracturas vertebrales en 62%, a los 36 meses en comparación con el placebo (1,5% frente a 3,9%)20.
Con respecto a la seguridad del denosumab, se ha propuesto que podría asociarse con un mayor riesgo de infecciones, ya que el RANKL también se expresa en los linfocitos B y T. Sin embargo, en los trabajos clínicos, los índices de mortalidad asociados con infecciones fueron similares en los tratados con denosumab y las mujeres asignadas a placebo21.
El tratamiento con denosumab está contraindicado en hipocalcemia, hipoparatiroidismo, cirugía de la glándula tiroides o de las paratiroides, malabsorción o resección del intestino delgado, ya que presentan mayor riesgo de hipocalcemia. En los pacientes que reciben agentes inmunosupresores o con antecedentes de inmunosupresión y en los pacientes que presentan infecciones graves en el contexto del tratamiento. También, está indicado el examen odontológico completo, antes del tratamiento con denosumab22,23.
Se ha descrito la osteonecrosis mandibular (ONM) asociada a denosumab en pacientes con osteoporosis tras una extracción dental, existiendo una asociación temporal entre el uso del fármaco y la ONM, así como entre la suspensión del mismo y la mejoría24.
Además, en varios de los casos descritos en la literatura, la ONM suele aparecer tras una exodoncia. Aunque entre los principales factores de riesgo para la ONM se ha encontrado el uso de glucocorticoides, mieloma múltiple, tratamiento con quimioterapia, bifosfonatos intravenosos y patología dental o exodoncia25.
Dentro de las ventajas que ofrece el denosumab se encuentra la ausencia de efectos secundarios gastrointestinales y la posibilidad de uso en pacientes con enfermedad renal crónica con TFG <30ml/min, con un control periódico de niveles de vitamina D y calcio26.
Inhibidores de la catepsina KOdanacatibSobre la base del concepto que la proteasa catepsina K tiene un papel importante en la degradación enzimática del hueso, el uso de inhibidores de la catepsina-K ha surgido como un nuevo enfoque terapéutico. La catepsina K es una enzima lisosomal clave de los osteoclastos maduros activados, inhibiendo la función de los osteoclastos, pero preservando su viabilidad27,28.
El odanacatib es, actualmente, el inhibidor más potente de la catepsina K y el más avanzado en investigación clínica. La biodisponibilidad oral de los inhibidores de la catepsina K depende de la formulación, siendo más alta que la biodisponibilidad oral de los bifosfonatos. Es metabolizada por enzimas del citocromo P450 y puede interactuar con otros medicamentos29,30.
Estudios en fase 1 de odanacatib, a dosis orales de 50mg y 100mg, una vez a la semana, reduce las concentraciones séricas del marcador de la resorción ósea telopéptido C-terminal del colágeno tipo 1 (CTXs) en 62%. La administración diaria de odanacatib (10mg) reduce las concentraciones séricas de CTXs en 81%31.
En un estudio en fase 2 (Estudio OCEAN), dosis orales semanales de odanacatib se evaluaron en 399 mujeres con osteoporosis posmenopáusica. Después de 24 meses, odanacatib (50mg) aumentó la DMO en la columna lumbar en 5,7% y en cadera total en 4,1%, en comparación con el placebo. La supresión de los marcadores de resorción ósea fueron dosisdependiente. Es de destacar que un subgrupo de 32 mujeres fueron sometidas a biopsias óseas, seguido por histomorfometría, arrojando como resultado una reducción modesta y transitoria de marcadores de formación ósea sin supresión de la tasa de formación. Las reacciones adversas con odanacatib eran similares a las de placebo, destacándose lesiones similares a esclerodermia32.
La extensión del estudio OCEAN durante 2 años revela que la inhibición de la catepsina K con odanacatib resultó en una disminución en la mayoría de los marcadores de resorción pero no disminuyó los marcadores de formación ósea. Esto se asoció con un aumento en la DMO; el efecto sobre los marcadores bioquímicos fue rápidamente reversible al suspender el tratamiento33.
El estudio fase III (LOFT), aleatorizado, ciego, controlado con placebo, con análisis preplaneado provisional para permitir la terminación anticipada si había diferencias significativas, tuvo 3 objetivos primarios que fueron determinar fracturas vertebrales y de cadera radiológicas y fracturas clínicas no vertebrales. Los objetivos secundarios incluyeron aumento en DMO, marcadores de recambio óseo y la seguridad y tolerabilidad, incluyendo histología ósea. Los participantes eran mujeres de 65 años o más, con una DMO T-score ≤–2,5 en la cadera o cuello femoral o con una fractura vertebral y un T-score ≤–1,5. Ellos fueron aleatorizados para tabletas de odanacatib o placebo34.
Después de un análisis intermedio planificado, un comité independiente de monitorización de datos recomendó detener el estudio antes de tiempo, debido a la eficacia robusta y un perfil favorable riesgo/beneficio34.
Con respecto al perfil de seguridad los eventos adversos más comunes reportados incluyen cefalea, síntomas influenza-like, odinofagia, trastornos en la alimentación, xerostomía y molestias abdominales35,36.
El odanacatib es un compuesto lipofílico con pobre solubilidad, la dosificación de grasa en la dieta aumenta la secreción de bilis que puede aumentar aún más la disolución del fármaco37. Estudios farmacocinéticos demostraron que comidas con alto contenido de grasa aumentan el área bajo la curva (AUC) del inhibidor de la catepsina K en aproximadamente un 110%. Un desayuno rico en grasas antes de la dosificación, para las dosis 25-300mg, produjo un aumento en las concentraciones plasmáticas de aproximadamente el doble en relación con el ayuno38,39.
El odanacatib es un sustrato de la CYP3A4 lo que implica riesgo de interacciones farmacológicas. Un estudio evidenció que el uso concomitante con esteroides (prednisona) no causa efectos significativos sobre la exposición de los sustratos del CYP3A4 sensibles in vivo a dosis terapéuticas. La administración concomitante de prednisona 10mg, una vez al día, no tuvo efecto sobre la farmacocinética de odanacatib 10 mg40.
Otro estudio evaluó la administración en conjunto con warfarina evidenciando que la farmacocinética y la farmacodinamia no fue afectada por la administración de dosis múltiples de odanacatib, lo que indica que este no es un inhibidor clínicamente importante de CYP 2C9, 3A4, 2C19 o 1A2. Al igual que lo reflejado en otro estudio en administración conjunta con digoxina41,42.
RelacatibActúa de manera no selectiva en catepsinas K, L y V. Ensayos clínicos con este fármaco se suspendieron, después de la fase I, debido a las interacciones fármaco-fármaco con la frecuencia medicamentos prescritos acetaminofén, ibuprofeno y atorvastatina43.
BalicatibInhibidor de la catepsina K altamente selectivo, pero no es tan selectiva en células debido a su alta concentración en los lisosomas44.
Estudios fase II se llevaron a cabo en sujetos con osteoartritis y osteopenia/osteoporosis. Se compararon 675 mujeres posmenopáusicas en un año con cuatro dosis orales diarias (5, 10, 25 o 50mg) contra placebo. El tratamiento con balicatib 25 y 50mg redujo marcadores de resorción ósea (CTXs 61%, NTXu 55%) y sin cambio en los marcadores de formación de hueso. Se asoció con un aumento de la DMO relacionada con la dosis, alcanzando el 4,5% en la zona lumbar y el 2,2% en toda la cadera con los 50mg de dosis semanal44.
A pesar de estos resultados, se suspendió debido a eventos adversos relacionados con la piel, incluyendo erupciones y esclerodermia like, lesión similar a la morfea45.
Inhibidores de Src quinasaSaracatinibLa Src quinasa es de la familia de la tirosina quinasa y es esencial para la función de los osteoclastos y la resorción ósea. Juega un papel en muchas de las vías de señalización responsables de la supervivencia de los osteoclastos, la motilidad y la activación por el RANKL46.
El saracatinib inhibe la Src quinasa y la Abl quinasa, implicada en la proliferación celular, la diferenciación y respuesta al estrés oxidativo, pero tiene muy poca actividad contra esta47.
Su efecto se investigó sobre el recambio óseo en hombres sanos. El estudio fue aleatorizado, doble ciego, controlado con placebo, de dosis múltiple ascendente, ensayo de fase I de saracatinib. Formaron parte del estudio 59 hombres sanos con edad media de 34,6 años, estos se dividieron en 5 cohortes; 4 con 12 sujetos y uno con 11 sujetos al azar, dentro de cada cohorte en la proporción de 3:1 para recibir una dosis única de saracatinib o placebo, respectivamente, seguidos de 7 a 10 días más tarde con dosis diarias durante otros 10 a 14 días. Los marcadores de recambio óseo se midieron antes de la dosis y a las 24 y 48 h después de la dosis única inicial e inmediatamente antes y 24 y 48 h y de 10 a 14 días después de la dosis final.
A una dosis de 250 mg (dosis máxima tolerada), el CTX disminuyó en 88% (IC 95% 84-91%) y NTXu disminuyó en 67% (IC 95% 53-77%) de la línea de base 24 h después de la dosis final. No hubo efecto significativo sobre los marcadores de formación ósea. No hubo eventos adversos significativos. Aunque la erupción papular se presentó en 30 vs. 6% y los pacientes presentaron diarrea en 24 frente a 0%, estas reacciones fueron más frecuentes en los hombres con saracatinib que en los que recibieron placebo48.
Se está probando actualmente en estudios fase 2 para tumores sólidos y metástasis óseas, pero no para la osteoporosis49.
Se llega, entonces, a la conclusión que la inhibición de Src quinasa reduce la resorción ósea osteoclástica en los seres humanos. El saracatinib es un tratamiento potencialmente útil para enfermedades caracterizadas por el aumento de la resorción ósea, tales como la enfermedad metastásica ósea y la osteoporosis50.
Análogos del péptido similar al glucagónEl péptido similar al glucagón (GLP-2) puede desacoplar el proceso de resorción ósea de la formación de hueso en la noche, es así como reduce resorción nocturna sin afectar la formación de hueso. Dado que el GLP-2 tiene una vida media en el plasma corta (7min) el efecto solo se presenta en la noche y la postprandial normal, reduciendo la resorción ósea en el desayuno. Esto podría cambiar la balanza de la remodelación a favor de la formación de hueso, dando como resultado un aumento neto en la DMO después de repetidas administraciones51.
El GLP-2 aplicado a las 10 p.m, en mujeres posmenopáusicas, durante 14 días, se traduce en una disminución dependiente de la dosis de la resorción ósea nocturna, según la evaluación de CTXs52.
Un ensayo terapéutico de 4 meses, doble ciego controlado con placebo, comparó 3 dosis diferentes de GLP-2 (0,4mg, 1,6mg y 3,2mg, administradas en la noche) contra una inyección de solución salina de control. Se evidenció aumento de la DMO en la cadera en 1,1% a partir de línea de base, pero no de la columna lumbar, no suprime ni estimula los marcadores de formación ósea. Las tasas generales de eventos adversos en los 4 grupos de tratamiento fueron similares y no hubo signos de taquifilaxia o anticuerpos contra el GLP-2. Los resultados indican que el GLP-2 produce una disminución sustancial en la resorción ósea sin supresión de la formación de hueso53.
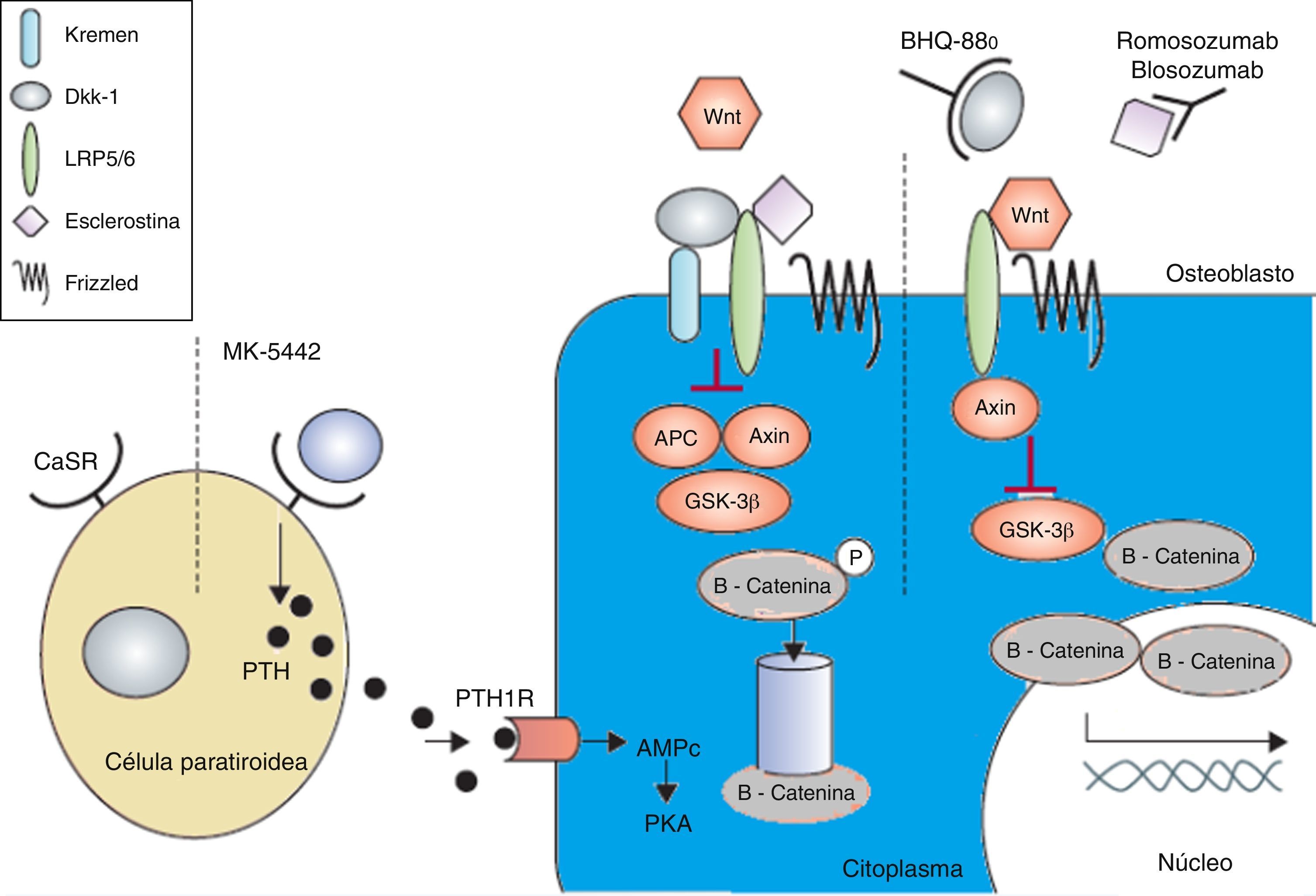
Terapia anabólicaLos fármacos anabólicos mejoran la formación de hueso en vez de evitar una mayor pérdida de masa ósea y el resultado es un aumento más rápido de la masa ósea. Se está prestando especial atención a la activación en la vía de señalización osteoformadora canónica Wnt para lograr un efecto anabólico en el hueso. La vía de señalización canónica Wnt ofrece varios objetivos que pueden ser adecuados para la intervención farmacológica. El principal objetivo de estas intervenciones es aumentar la señalización con el fin de aumentar la masa ósea (figs. 2 y 3). Esto se ha logrado en modelos animales con la inhibición de dickkopf-1 (DKK-1), la glucógeno sintasa quinasa-3b (GSK-3b), o esclerostina, como se describe a continuación54,55.

El receptor sensible al calcio se antagoniza por MK-5442 y desencadena explosiones cortas de secreción de PTH. La unión de PTH a su receptor mejora las funciones de los osteoblastos y la formación de hueso. La presencia de antagonistas de Wnt - DKK-1 y esclerostina inhiben la señalización. El DKK-1 se necesita para formar complejo con Kremen y posteriormente se une a LRP5/6, mientras que la esclerostina se une directamente a LRP5/6. El BHQ-880 y AMG-785 son anticuerpos de DKK-1 y antiesclerostina respectivamente. Después de neutralizar el DKK-1 y la esclerostina, el Wnt puede unirse al LRP5/6, lo que resulta en la degradación de GSK-3. Como consecuencia la -catenina es estabilizada, se acumula y se transloca al núcleo donde regula la transcripción de genes osteoblásticos.
AMPc:adenosina monofosfato cíclico; APC: adenomatosis poliposis coli; CaSR:receptor sensible al calcio; DKK-1:dickkopf-1; GSK:proteína relacionada con el receptor glucógeno sintasa quinasa 3; PKa:proteína kinasa A; PRL:lipoproteína de baja densidad; PTH:hormona paratiroidea; PTH1R:receptor PTH1.
Adaptada y modificada de Rachner TD, Khosla D, Hofbauer LC. Osteoporosis: Now and the future. Lancet. 2011; 377: 1276-87.

La lipoproteína de baja densidad (LRP5) puede formar un complejo con DKK-1, que desencadena una rápida internalización y agotamiento de LRP5, dando lugar a la inhibición de la vía canónica de señalización Wnt.
Los estudios genéticos con ratones que carecen de un solo alelo de DKK-1 mostraron un marcado aumento de volumen de hueso trabecular y tasa de formación ósea56.
La neutralización de DKK-1 es aún limitada en los ensayos preclínicos. El bloqueo de DKK-1 inhibe la pérdida de hueso en un modelo de artritis reumatoide y de mieloma múltiple, la inhibición de DKK-1 impide la formación de lesiones osteolíticas y aumenta la tasa de formación ósea.
Actualmente, se está investigando en pacientes con mieloma múltiple refractario. Sin embargo, los efectos de neutralización de DKK-1 todavía no se han investigado en la osteoporosis57.
Inhibidores heterocíclicos de la glucógeno sintasa quinasa-3bLa inhibición de la GSK-3b podría prevenir la fosforilación de b-catenina, lo que conduce a la estabilización de interacciones del complejo b-catenina con Wnt58.
Los ratones tratados con cloruro de litio como un inhibidor de GSK-3 indicaron un aumento en la formación de hueso y la masa ósea. El tratamiento de ratas ooforectomizadas con un inhibidor activo por vía oral de GSK dual a/b, durante 2 meses resultó en un aumento en el número de trabéculas así como en el área trabecular y el grosor. Se incrementó la DMO en los sitios de hueso esponjoso y esto se asoció con el aumento de la formación de hueso en histomorfometría59.
Anticuerpos monoclonales antiesclerostina (romosozumab - blosozumab)La esclerostina es un producto del gen Sost que inhibe la osteoblastogénesis. Las mutaciones del Sost resultan en expresión ausente de la esclerostina y es responsable de esclerosteosis y de la enfermedad de van Buchem. Ambas displasias esqueléticas que se caracterizan por marcados aumentos en la masa ósea60.
Los anticuerpos monoclonales humanizados antiesclerostina causan mejoría de la señalización del Wnt y un aumento de la masa ósea en roedores y primates no humanos. En un modelo en ratas posmenopáusicas con osteoporosis debido a ooforectomía, el tratamiento con un anticuerpo monoclonal antiesclerostina aumentó la masa ósea e impidió la pérdida ósea asociada con la deficiencia de estrógenos61.
En una etapa posterior en humanos, se demostró que los anticuerpos antiesclerostina pueden aumentar la DMO y los marcadores bioquímicos de formación ósea en los seres humanos61.
En un estudio fase 1, aleatorizado, doble ciego, de dosis única controlado con placebo, 72 sujetos sanos recibieron AMG 785 o placebo por vía subcutánea (0,1 - 0,3 - 1 - 3 - 5 - 10mg/kg) o por vía intravenosa (1 o 5mg/kg) y fueron seguidos durante un máximo de 85 días, fue bien tolerado y aumentó los marcadores de formación ósea entre 60-100% al día 21.
Hubo un aumento estadísticamente significativo en la DMO de hasta 5,3% en la columna lumbar y un 2,8% en toda la cadera en comparación con el placebo61.
El romosozumab aumentó el propéptido aminoterminal tipo 1 de 66 a 147%, disminución del C-telopéptido sérico (sCTX) de 15 a 50%, y aumentó la DMO de columna lumbar entre 4-7%. Las tasas de eventos adversos fueron equilibradas entre los grupos sin hallazgos significativos de seguridad. Estos datos apoyan la inhibición de esclerostina en los trastornos que podrían beneficiarse de una mayor formación ósea62.
Un estudio de fase II, multicéntrico, aleatorizado, controlado con placebo, paralelo, de 8 grupos se realizó durante un período de 12 meses en 419 mujeres posmenopáusicas, con edades de entre 55 y 85 años con baja DMO (T-score entre –2,0 y –3,5 en la columna lumbar, cadera total o cuello femoral). Las participantes fueron asignadas aleatoriamente a uno de los 8 grupos: placebo cada 3 meses o mensual, romosozumab 140mg SC cada 3 meses, romosozumab 210mg SC cada 3 meses, romosozumab 70mg SC mensual, romosozumab 140mg SC mensual, romosozumab 210mg SC mensual, alendronato 70mg a la semana y teriparatide 20 mcg SC diario. Las participantes fueron seguidas durante 12 meses, la DMO fue evaluada al inicio y a los 3, 6 y 12 meses y se realizaron laboratorios al inicio del estudio, a la semana 1, y a los meses 1, 2, 3, 6, 9 y 12. De las 419 participantes inscritas en el estudio, 383 completaron la visita de 12 meses, mientras que 36 se retiraron. La variable principal del estudio fue el cambio porcentual desde el inicio en la DMO en la columna lumbar en el mes 12, y los objetivos secundarios incluyeron el cambio porcentual desde el inicio en la DMO en la cadera total, cuello femoral y tercio distal del radio en el mes 12. En comparación con el placebo, todos los grupos tratados con romosozumab tuvieron aumentos significativos en la DMO en la columna lumbar, cadera total y cuello femoral a los 12 meses. Las mayores ganancias se observaron con dosis mensual de romosozumab (210mg) con un aumento de 11,3% en la DMO lumbar, aumento de 4,1% en el total de la DMO de cadera y el aumento de 3,7% de la DMO en el cuello femoral a los 12 meses, que fueron mayores que el aumento visto en las tratadas con teriparatide y alendronato. No hubo diferencias significativas en el tercio distal del radio a los 12 meses en cualquier grupo. A los 6 meses, la DMO de columna lumbar y cadera total se incrementaron significativamente en todos los grupos tratados con romosozumab, en comparación con el tratamiento con placebo, mientras que la DMO del cuello femoral fue mayor en los grupos que recibieron 140mg romosozumab mensual, 210mg mensuales o 210mg cada 3 meses, en comparación con el tratamiento con placebo63.
Marcadores de recambio óseo también se midieron como criterios de valoración secundarios en los meses 1, 3, 6, 9 y 12. En todos los grupos romosozumab, el marcador de la resorción ósea, CTXs, cayó desde la línea de base, con la mayor disminución en la primera semana, y en los grupos que recibieron dosis mensuales de romosozumab, se mantuvo por debajo de la línea de base en el mes 1263.
El tratamiento con romosozumab se asoció con una disminución dependiente de la dosis en los niveles de calcio de 1,30 a 2,68% desde el inicio, con un nadir en el mes uno y un aumento compensatorio en PTH. El calcio sérico regresó a la línea de base en las visitas de seguimiento y no hubo eventos adversos asociados con el cambio de valor de laboratorio. No hubo diferencia en la proporción de participantes que informaron eventos adversos entre el grupo de placebo (90%) y los grupos romosozumab (87%), y no hubo ninguna relación aparente entre la dosis. Sin embargo, reacciones en el lugar de inyección fueron más frecuentes con romosozumab que con placebo63,64.
Con base en los resultados prometedores observados en los estudios iniciales, hay varios de fase adicional I y II, los estudios que se han completado o están en curso, incluyendo un estudio de evaluación de la seguridad de romosozumab en pacientes con enfermedad renal crónica en fase terminal y estudios de evaluación de los efectos de romosozumab en la curación de fracturas. Además, hay 5 estudios en curso de fase III, 4 que investigan el uso de romosozumab en mujeres posmenopáusicas con osteoporosis y uno que investiga el uso de romosozumab en hombres con osteoporosis65.
Si bien la investigación de romosozumab continúa, se están desarrollando otros anticuerpos monoclonales humanizados contra esclerostina. Más específicamente, blosozumab se ha investigado en estudios fase I y estudios de fase II. En el estudio fase I, realizado en mujeres posmenopáusicas sanas, los investigadores encontraron que hubo respuestas dosis-dependientes en esclerostina, P1NP, BSAP, osteocalcina, CTXs, y la DMO con dosis únicas y múltiples de blosozumab. Al día 85, hubo hasta un aumento de 3,41% y un aumento de hasta 7,71% del inicio en DMO de columna lumbar en la dosis única y múltiples dosis, respectivamente66.
En el estudio de fase II, 120 mujeres posmenopáusicas con una DMO baja recibieron blosozumab o placebo SC en dosis variables, durante 12 meses. Al final del estudio, la DMO en la columna lumbar aumentó en 17,7% y la DMO en la cadera total aumentó 6,2% en el grupo de dosis más alta. Además, los marcadores de formación ósea aumentaron inicialmente y luego tendieron hacia los niveles previos al tratamiento, mientras que los marcadores de resorción ósea permanecieron disminuidos. En general, el fármaco fue bien tolerado, por lo tanto blosozumab también se muestra prometedor como tratamiento para la osteoporosis67.
Sobre la base de nuestra comprensión de la fisiología de esclerostina y datos de estudios en humanos, hay varias posibles complicaciones y efectos adversos a considerar. En primer lugar, los pacientes con esclerostosis y la enfermedad van Buchem desarrollan engrosamiento del cráneo y huesos de la cara, lo que lleva a atrapamiento de los nervios craneales; estos pacientes también están en riesgo para el desarrollo de estenosis espinal. Podría especularse que las complicaciones similares podrían ocurrir con el tratamiento a largo plazo, debido a la acumulación excesiva de hueso en ubicaciones no deseadas. Sin embargo, la regulación positiva de esclerostina puede regular la ganancia de masa ósea como sugiere Stolina et al., y esto puede evitar la acumulación de masa ósea en exceso68. Tal efecto, sin embargo, queda por determinar en los seres humanos. Los estudios fase I y II han demostrado que romosozumab es generalmente bien tolerado con efectos adversos leves. Sin embargo, en el estudio de fase I, un paciente desarrolló hepatitis transitoria. No se sabe si esto fue una relación causal con el fármaco. Una disminución en el calcio sérico fue vista con la dosificación inicial. Los pacientes tuvieron aumentos compensatorios en la PTH y los niveles séricos de calcio regresaron a la línea de base, estas disminuciones en el calcio sérico podrían llegar a ser clínicamente significativas en los pacientes con deficiencia de vitamina D o enfermedad renal crónica69.
Tanto en estudios fase I y II algunos sujetos desarrollaron anticuerpos neutralizantes. Si bien esto no parece afectar a la farmacocinética o la farmacodinamia del fármaco, con el tratamiento a largo plazo, podría afectar la eficacia y la potencia del medicamento69.
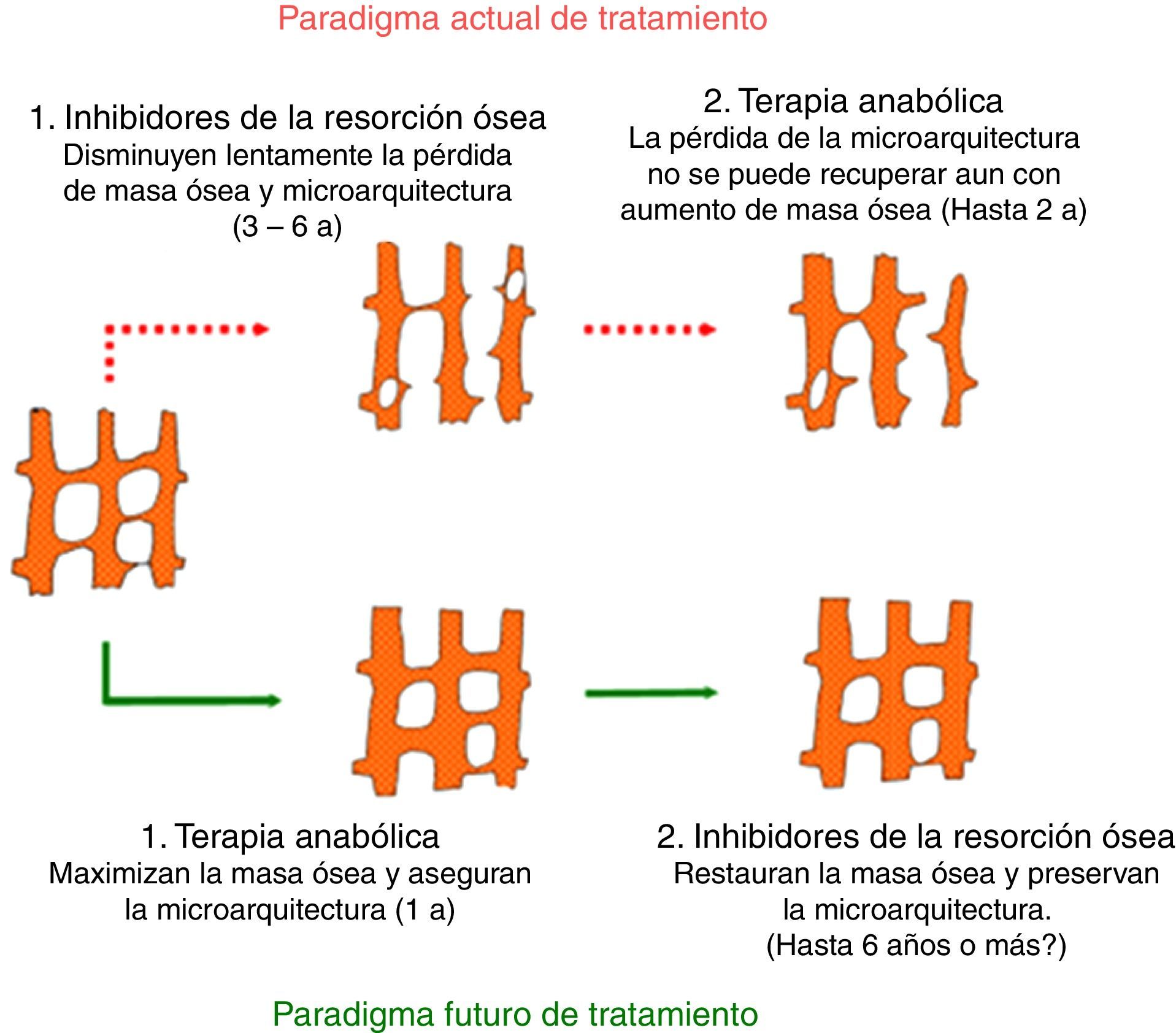
ConclusionesEl número de fármacos disponibles antiosteoporóticos aumentará considerablemente en los próximos años. Muchos de los nuevos medicamentos combinan eficacia con la administración conveniente, que podría traducirse en una mejor adherencia. El odanacatib y hasta cierto punto el saracatinib representan una clase distinta de antirresortivos que inhiben la actividad de los osteoclastos en lugar de menoscabar la viabilidad de estos. La integración con éxito de estos nuevos compuestos se basa en la evidencia de la terapia de la osteoporosis que requiere herramientas sencillas y aplicables para la toma de decisiones clínicas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
