



Reportar las diferentes manifestaciones clínicas y los esquemas terapéuticos instaurados en 3 pacientes con poliarteritis nudosa cutánea. Así mismo, describir la fisiopatología, el cuadro clínico, los criterios diagnósticos y las opciones terapéuticas.
MétodosSe realizó una búsqueda de la literatura en Google Académico y PubMed utilizando términos MeSH. En la selección de la bibliografía, la búsqueda no se limitó por fecha de publicación ni por idioma, debido al escaso número de reportes.
ResultadosLa poliarteritis nudosa cutánea es una vasculitis de pequeño y mediano vaso, infrecuente, poco reportada en Colombia. Su fisiopatología no es del todo conocida, se considera mediada por el depósito de inmunocomplejos en las paredes de vasos sanguíneos; puede ser desencadenada por agentes infecciosos y asociarse con enfermedades autoinmunes. Las manifestaciones cutáneas incluyen principalmente nódulos subcutáneos dolorosos, livedo reticular y úlceras. A nivel extracutáneo podrían encontrarse mialgias, artralgias, neuropatía periférica y fiebre. El diagnóstico representa un desafío. El tratamiento no está estandarizado y es guiado según la severidad de la patología.
ConclusiónLa poliarteritis nudosa cutánea es una entidad poco frecuente, con una amplia variedad de manifestaciones cutáneas. Hasta el momento, no contamos con una prueba diagnóstica específica. Su diagnóstico representa un reto para el dermatólogo y requiere un manejo multidisciplinario, en el cual el reumatólogo desempeña un papel fundamental.
To report the clinical manifestations and therapeutic schemes established in 3patients with cutaneous polyarteritis nodosa, as well as to describe the pathophysiology, clinical features, diagnostic criteria, and therapeutic options.
MethodsA literature review was performed using Google scholar and PubMed and MeSH terms. There was no limit into the publication date or language for the selection of the articles.
ResultsCutaneous polyarteritis nodosa is an uncommon small and medium-sized vessel vasculitis, and is rarely reported in Colombia. Although its pathophysiology is unknown, it is considered to be mediated by the deposition of immunocomplexes in the walls of blood vessels. It can be triggered by infectious agents and be associated with autoimmune diseases. The cutaneous manifestations mainly include subcutaneous painful nodules, livedo reticularis, and ulcers. Myalgia, arthralgia, peripheral neuropathy, and fever can also be present. It represents a diagnostic challenge. Treatment is not standardised and is guided according to the severity.
ConclusionCutaneous polyarteritis nodosa is a rare entity, with a wide variety of cutaneous manifestations. There is still no specific diagnostic test. Its diagnosis represents a challenge for the dermatologist, and requires multidisciplinary management, in which the rheumatologist plays a fundamental role.
La poliarteritis nudosa (PAN) es una vasculitis sistémica necrosante que afecta a arterias de pequeño y mediano calibre, principalmente en el riñón, en el sistema nervioso periférico y en la piel, sin cursar con compromiso pulmonar, según los registros clínicos clásicos1-3. Se considera un diagnóstico de exclusión frente a otros tipos de vasculitis de mediano y pequeño calibre y se caracteriza por presentar anticuerpos contra citoplasma de neutrófilos (ANCA) negativos4. La PAN puede comprometer la piel de manera aislada, caso en el cual se denomina PAN cutánea (PANc), variante clínica de presentación poco frecuente y etiología aún no dilucidada1. La PANc representa entre el 4 y el 10% de todos los casos de PAN1,5. A continuación, se reportan 3 casos confirmados por enfermedad que conllevaron un reto diagnóstico para el personal médico a cargo y requirieron diferentes esquemas terapéuticos, debido a su curso clínico y severidad variable. Finalmente, se hace una breve revisión de esta entidad.
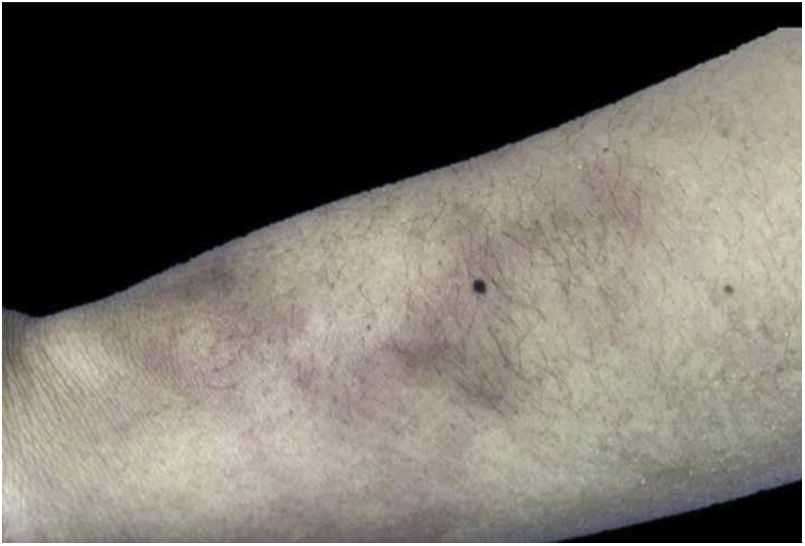
Casos clínicosCaso 1Mujer de 34 años, publicista cesante, que consultó por 3 años de nódulos eritematosos, subintrantes y dolorosos a la palpación, localizados en tórax, dorso y miembros inferiores. Describía el dolor como lancinante, incapacitante, habitualmente de 8/10 en escala visual análoga. A la revisión por sistemas refería artralgias intermitentes de baja intensidad en codos, rodillas y tobillos, de predominio vespertino, sin deformidad ni rigidez, así como mialgia en antebrazos proximal, en muslos distal y en piernas a nivel de gastrocnemios, que se acentuaban con la palpación, sin debilidad asociada en segmentos proximales o distales.
Dentro de los antecedentes personales, refería que 2 años atrás había recibido tratamiento extrainstitucional con prednisolona y metotrexato (dosis y frecuencia no recordadas por la paciente ni registradas en la historia clínica), así como ciclofosfamida 6g por vía intravenosa (IV) en total, ante impresión diagnóstica de miopatía inflamatoria no específica, sin mejoría de las lesiones ni síntomas relacionados.
En el examen físico, además de las lesiones en piel (fig. 1), se puso en evidencia comunicación de ambas fosas nasales a través del tabique nasal, en su porción cartilaginosa, dolorosa a la palpación, compatible con perforación septal anterior. Debido a este hallazgo clínico, se llevaron a cabo estudios de extensión que permitieron descartar granulomatosis con poliangitis y otras vasculitis primarias sistémicas, a partir de estudios por imagen de pulmón y de función renal, además de uroanálisis y morfología eritrocitaria en muestra de orina ocasional, excluyendo hematuria microscópica de origen glomerular.

Se solicitaron los siguientes paraclínicos: Elisa virus de la inmunodeficiencia humana (VIH), hepatotropos, crioaglutininas, anticuerpos antinucleares (ANA), anticuerpos nucleares extractables (anti-ENA), ANCA, factor reumatoide (FR), anticuerpos antipéptidos citrulinados (anti-CCP), así como anticuerpos antifosfolípidos (anticoagulante lúpico, anticardiolipina IgM e IgG, anti B2-glicoproteína IgM e IgG), que fueron negativos. De igual forma, la paciente negaba en la anamnesis traumatismo facial reciente (que explicara reabsorción de hematoma septal) o consumo de alcaloides inhalados.
A continuación, se tomaron 6 baciloscopias de linfa, las cuales fueron negativas para Mycobacterium leprae, y una biopsia de piel en la que no se evidenciaron granulomas ni infiltrado dérmico difuso por histiocitos, compromiso neural ni granulomatoso, con coloración de Fite-Faraco negativa; de igual forma, se descartó leishmaniasis como diagnóstico diferencial asociado. La biopsia de piel documentó paniculitis mixta de predominio septal, con vasculitis de mediano y pequeño vaso.
Los estudios de electromiografía y velocidad de conducción fueron negativos para mononeuritis múltiple o variantes de polineuropatía simétrica. Se estimó como diagnóstico de exclusión PANc, por lo que se inició prednisolona a 1mg/kg/día en esquema de titulación progresiva por 4 semanas, sin lograr mejoría clínica.
Ante la persistencia de las lesiones se remitió a Reumatología y se continuó con prednisolona a 25mg/día, con inicio de metotrexato a 15mg/semana. Por la pobre respuesta y el mal control del dolor, la paciente recibió 2 dosis adicionales de ciclofosfamida (500mg IV cada una, cada 15 días). Sin embargo, siguieron apareciendo nuevas lesiones en superficies extensoras, por lo que se decidió el inicio de infliximab, a una dosis de 5mg/kg. Hasta el momento existe evidencia de haber completado 3 dosis en total (semanas 0, 2 y 6), con disminución en la intensidad del dolor y el tamaño de las lesiones cutáneas; queda pendiente continuar igual dosis acordada cada 8 semanas, lo que se propuso como fase de mantenimiento.
En cuanto a la perforación septal anterior, no hay claridad acerca del tiempo de aparición y la causa precisa de esta. A la fecha, se han descartado las siguientes etiologías: poliangitis con granulomatosis, policondritis recidivante, consumo ocasional de cocaína inhalada, hematoma septal resuelto, leishmaniasis mucosa y enfermedad de Hansen. En la actualidad, se encuentra en seguimiento ambulatorio por Otorrinolaringología.
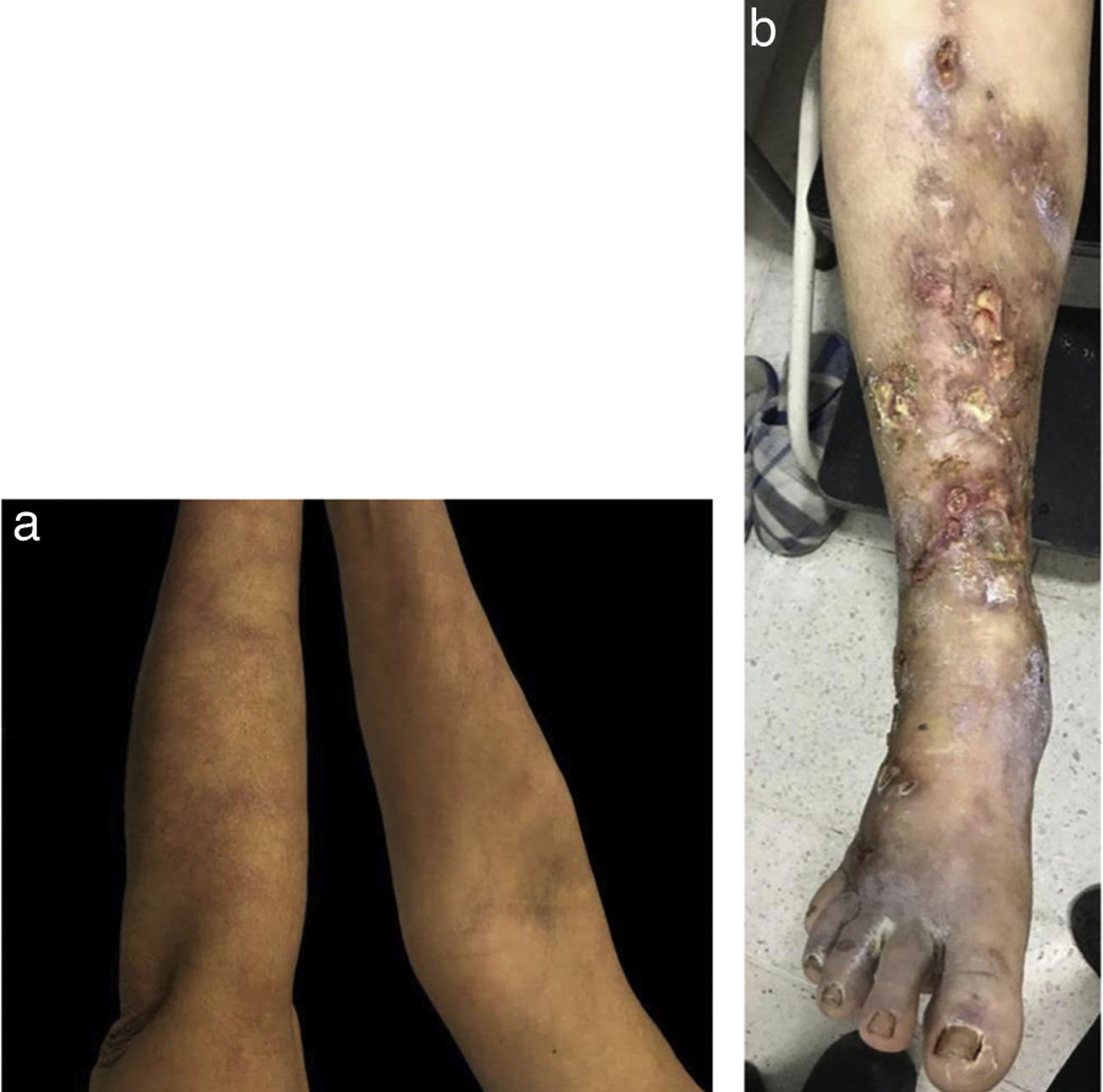
Caso 2Mujer de 58 años, ama de casa, sin antecedentes personales o familiares de importancia, que consultó por cuadro clínico de 8 años de evolución de nódulos eritematosos en miembros inferiores, dolorosos, los cuales se habían ulcerado en los últimos 3 meses, asociándose con secreción purulenta y dolor intenso de tipo lancinante. Había recibido tratamiento con antibióticos tópicos (ácido fusídico, mupirocina) y múltiples curaciones con sulfadiazina de plata, sin mejoría clínica. Refería además mialgias en miembros inferiores, sin debilidad, artralgias/artritis o signos de derrame articular, sin rigidez o déficit motor asociado.
En el examen físico presentaba, de forma generalizada, lesiones purpúricas de configuración reticular (fig. 2A y B), en la cara anterior de las piernas, los tobillos y el dorso de pies, úlceras con bordes regulares definidos y otras con borde estrellado confluyentes, además de áreas de atrofia blanca en la periferia, dolorosas, que limitaban la marcha. La impresión diagnóstica inicial fue PAN clásica.

Se procedió a realizar una biopsia de piel, la cual reportó una vasculitis de pequeño y mediano vaso, sin formación de granulomas. En la inmunofluorescencia directa se observaron depósitos perivasculares de IgM, C1q y C3.
Paralelamente, los estudios para descartar autoinmunidad (ANA, anti-ENA, ANCA, FR, anti-CCP, anticoagulante lúpico, anticardiolipina IgM e IgG, anti-B2-glicoproteína IgM e IgG) fueron negativos. De igual forma, se descartó compromiso sistémico de tipo pulmonar, renal, gastrointestinal y en el sistema nervioso central y periférico. Ante la negatividad del perfil de autoinmunidad, se descartó el diagnóstico de lupus eritematoso sistémico y se determinó por exclusión el diagnóstico de PANc.
Se indicó tratamiento inmunosupresor con prednisolona 1mg/kg/día por vía oral, además de ampicilina/sulbactam, por hallazgos clínicos indicativos de sobreinfección bacteriana. La paciente mejoró progresivamente durante la hospitalización y se le dio egreso con prednisolona en esquema de desmonte convencional, hasta dejarla con dosis de mantenimiento de 7,5mg/día y orden para nueva valoración por parte de Reumatología. Se perdió el seguimiento clínico debido a aspectos sociodemográficos, sin que a la fecha se conozca su evolución.
Caso 3Hombre de 27 años, fotógrafo, que acudió a consulta médica por cuadro de 3 meses de lesiones ulcerativas en los pies y la región plantar, precedidas de cianosis acral intensificada con las bajas temperaturas.
Como antecedentes personales refería artritis idiopática juvenil, diagnosticada a sus 5 años, que requirió tratamiento con metotrexato y desde los 18 años etanercept durante 2 años (a dosis y esquemas desconocidos) y que suspendió a los 20 años por decisión propia. Adicionalmente, diagnóstico de infección por VIH desde los 25 años, en el momento clasificado como estadio 2, con adherencia del 100% a la terapia antirretroviral (efavirenz, emtricitabina y tenofovir) y adecuado estado inmunovirológico (carga viral indetectable, recuento de linfocitos CD4+mayor de 500 células/mm3), sin evidencia de infecciones oportunistas en historia clínica.
En la revisión por sistemas, refería hipoestesias con ambos pies con patrón «en calcetín», sin artralgias ni efusión articular, con marcha antálgica secundaria.
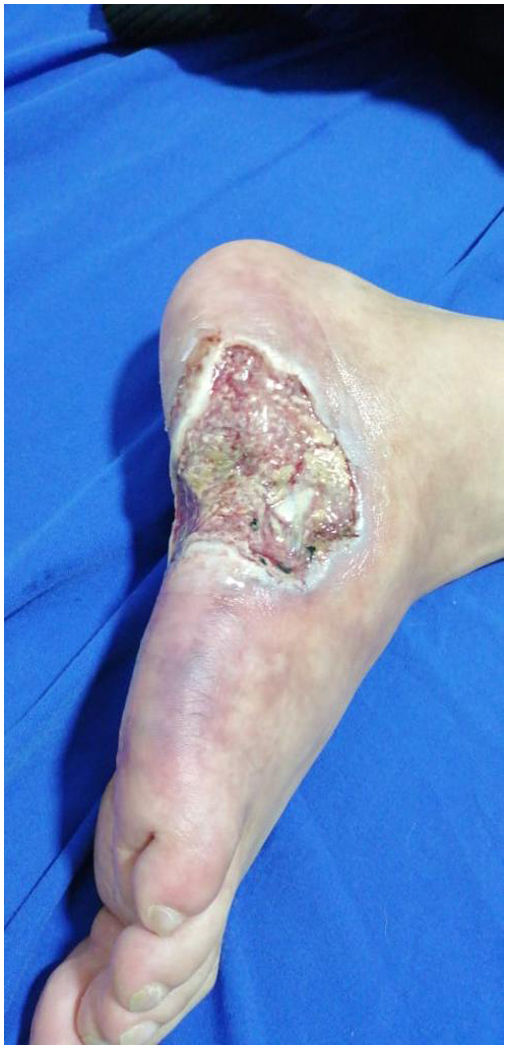
En el examen físico presentaba máculas violáceas reticuladas de distribución irregular e incompleta sobre el dorso y la cara lateral del pie derecho; hacia la cara lateral del pie tenía una úlcera de 6×4cm, de bordes macerados e irregulares, dolorosa a la palpación, de fondo limpio, con exposición de tendones y algunos nódulos eritematosos y violáceos perilesionales menores de 1,5cm (fig. 3), sin artritis, dactilitis o entesitis, ni tampoco dolor lumbar de características inflamatorias. Pulsos distales palpables en extremidades inferiores (femoral, pedio, tibial posterior, poplíteo), sin signos de isquemia crítica.

Debido al aceptable control inmunológico en el momento de la consulta, así como la exclusión de trastornos vasculares relacionados, entre los diagnósticos diferenciales se consideraron pioderma gangrenoso y vasculitis en el escenario de infección por retrovirus, tanto de etiología infecciosa (vasculitis asociada a citomegalovirus [CMV], Mycobacterium tuberculosis [M. tuberculosis] y virus de la hepatitis B [VHB] y C [VHC], así como por el mismo VIH, usualmente relacionada con depósitos de IgA) como asociada con inmunocomplejos (vasculitis por hipersensibilidad y vasculitis inmunoproliferativa angiocéntrica). Se realizaron paraclínicos complementarios: crioaglutininas, función renal, electrólitos, así como estudios para evaluar autoinmunidad (ANA, anti-ENA, ANCA, FR, anti-CCP), y anticuerpos antifosfolípidos (anticoagulante lúpico, anticardiolipina IgM e IgG, anti B2-glicoproteína IgM e IgG), que fueron negativos.
Así mismo, se llevó a cabo una tomografía computarizada (TAC) de tórax y abdomen, simple y con contraste, sin evidencia de hallazgos significativos; no se complementó con evaluación angiográfica de vasos abdominales. Finalmente, los estudios electrofisiológicos complementarios descartaron lesión de nervio periférico.
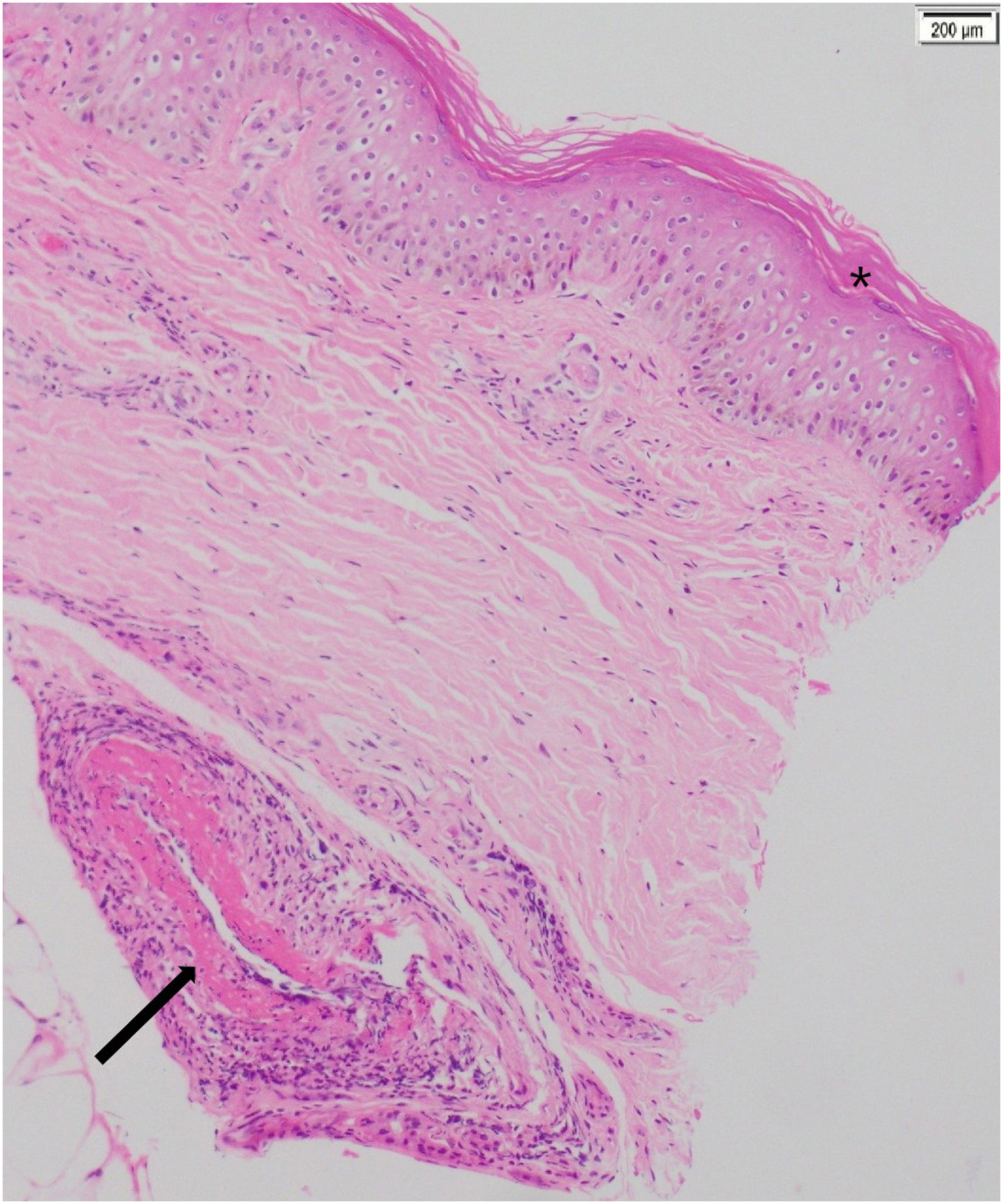
Se realizó una biopsia de piel, que documentó una vasculitis de pequeño y mediano vaso e inflamación aguda abscedada, sin formación de granulomas, con coloraciones para hongos y micobacterias negativas (fig. 4).
. Hematoxilina y eosina, 10x. Epidermis con ortoqueratosis compacta (*). En la dermis profunda, en cercanía del tejido celular subcutáneo, se observa vasculitis de un vaso de mediano calibre con leucocitoclastia e hiperproliferación de la capa íntima, sin trombos intraluminales (flecha).")
Biopsia de piel (paciente número 3). Hematoxilina y eosina, 10x. Epidermis con ortoqueratosis compacta (*). En la dermis profunda, en cercanía del tejido celular subcutáneo, se observa vasculitis de un vaso de mediano calibre con leucocitoclastia e hiperproliferación de la capa íntima, sin trombos intraluminales (flecha).
Se consideró que la artritis idiopática juvenil se encontraba en remisión, debido a la ausencia de síntomas o signos clínicos de actividad, sin medicamentos que pudieran generar estos hallazgos clínicos. Como opción diagnóstica más probable se consideró PANc, descartándose como principal diagnóstico diferencial PAN -like asociada con infección por VIH, vasculitis secundaria que se puede presentar con cualquier conteo de linfocitos CD4+.
A continuación, se indicó tratamiento con prednisolona 1mg/kg/día (50mg/día) y azatioprina 50mg/día vía oral, con notable mejoría clínica a las 4 semanas de iniciado el tratamiento. Posteriormente, se inició descenso de dosis del esteroide por esquema convencional hasta 20mg/día y se continuó con la dosis de azatioprina ya señalada. El paciente, que aún se encuentra en seguimiento por parte del servicio de dermatología, ha logrado resolver la marcha antálgica y presenta avances en el proceso de cicatrización de sus lesiones.
DiscusiónLa PAN fue la primera vasculitis sistémica descrita en la literatura médica. Hacia 1866, Kussmaul y Maier caracterizaron esta condición a partir de los hallazgos anatomopatológicos de una vasculitis necrosante de medianas y pequeñas arterias (excluyendo arteriolas, capilares y vénulas)1,2. Existen 3 variantes clínicas descritas para esta entidad: la forma clásica idiopática, la asociada con infección por VHB y la PANc. Esta última, descrita por Lindberg en 1931 como una vasculitis de vasos de mediano calibre limitada a piel3,5-7, puede evolucionar a PAN clásica, en cuyo caso cambiaría el pronóstico y el tratamiento. Según el último consenso de Chapel-Hill del 2012, esta variante se encuentra reclasificada como una arteritis de órgano único (en este caso, la piel)6-8.
No existen reportes de prevalencia o incidencia de PANc debido a su infrecuente presentación. Un estudio que incluyó 79 casos de PANc encontró una relación mujer:hombre de 1,7:14,6. La PANc afecta principalmente a mujeres mayores de 40 años, con un pico entre 50-59 años5,8,9. En el registro de Nakamura et al.10, con 22 pacientes entre 17-77 años, el 86% eran mujeres. Lo anterior se correlaciona con nuestro reporte, en el cual 2 de los 3 casos fueron mujeres, ambas mayores de 40 años. En la tabla 1 se incluyen los reportes de caso documentados en Colombia a la fecha11-14.
Registros de PAN cutánea (PANc) en Colombia
| Iglesias et al.11Serie de casos | Olmos et al.12Reporte de caso | Quintana et al.13Reporte de caso | Cárdenas et al.14Reporte de caso | Ordóñez et al. (actual)Reporte de caso | |
|---|---|---|---|---|---|
| Distribución por género (número de casos) | 4 casos: mujer (3), hombre (1) | Hombre (1) | Mujer (1) | Mujer (1) | 3 casos: mujer (2), hombre (1) |
| Edad promedio | 18-44 años | 19 años | 36 años | 58 años | 27-58 años |
| Manifestaciones clínicas | |||||
| Nódulos eritematosos (localización) | +(miembros superiores e inferiores) | +(miembros superiores e inferiores) | +(muslo, seno derecho, antebrazos, cuero cabelludo y tercio superior de la pierna derecha) | +(miembros inferiores) | +(tórax, dorso [1] y miembros superiores e inferiores) |
| Fiebre | + | + | – | – | – |
| Mialgias | + | + | – | – | +(miembros superiores e inferiores) (2) |
| Artralgias (localización) | +(rodillas y tobillos) | + | – | – | +(codos, rodillas y tobillos) (2) |
| Livedo reticular | – | +(cuello, región sacra y extremidades) | – | +(miembros inferiores) | +(miembros inferiores) (2) |
| Úlceras | +(miembros superiores e inferiores) | – | – | +(miembros inferiores) | +(miembros inferiores) (2) |
| Hallazgos patológicos (biopsia de piel)(número de casos) | Vasculitis leucocitoclástica de arteriolas y arterias de mediano calibre con necrosis fibrinoide | Vasculitis leucocitoclástica de arteria de mediano calibre, con trombosis y oclusión total de su luz por depósito de fibrina | Vasculitis leucocitoclástica de arterias de mediano calibre con necrosis fibrinoide | Vasculitis leucocitoclástica de arteriolas y arterias de mediano calibre con necrosis fibrinoide | Paniculitis mixta de predominio septal (1), vasculitis de mediano y pequeño vaso, sin granulomas (3) |
| Inmunofluorescencia directa | Depósito de Ig M, G y C3 en la pared de los vasos | – | – | – | Depósitos de IgM, C1q y C3 en la pared de los vasos (1) |
| Tratamiento(número de casos) | Prednisona 30-60 mgAzatioprina 100 mg/díaCiclofosfamida 100mg VO diario+prednisona | Prednisona 0,5 mg/kg/día | Prednisona 15-30 mg/díaAzatioprina 100 mg/díaColchicina 1 mg/díaCiclofosfamida, 500mg IV mensual×2 dosis | Prednisolona (no describen dosis)Ciclofosfamida 500mg IV mensual, por 2 años | Prednisolona 1 mg/kg/día (2) posterior 20-25 mg/día (2)Metotrexato 15 mg/ semanales (1)AzatioprinaDos dosis de ciclofosfamida de 500mg IV (1)Infliximab 5 mg/kg/dosis semana 1, 2, 6 y cada 8 semanas |
La etiología de la PANc es desconocida1-5, se cree que es un desorden mediado por inmunocomplejos, con depósito de C3 e IgM en la pared de los vasos sanguíneos8-10. Agentes infecciosos como los virus hepatotropos (B y C) se han asociado con su inicio o recurrencia2,10,15, por lo que es obligatorio realizar serología para estos virus en pacientes con sospecha de esta condición clínica, tal como se hizo en los casos presentados.
Criado et al.16 reportaron a M. tuberculosis como el agente infeccioso más comúnmente asociado, seguido por el CMV. El mecanismo más aceptado es la estimulación de linfocitos T y B autorreactivos, ya que las tinciones de Ziehl-Neelsen han sido negativas para detectar presencia de micobacterias en biopsias de piel.
En países en vía de desarrollo, la PANc se ha asociado con infección por el VIH15,17. Si bien el VIH propiamente dicho puede desencadenar una vasculitis que no es exclusiva de pequeño tamaño15, en el caso 3 el paciente cumplía características clínicas e histológicas de PANc, habiéndose descartado otro tipo de vasculitis, por lo que se consideró un epifenómeno inmunológico asociado con la enfermedad infecciosa vírica de base.
Entre causas no infecciosas se ha descrito asociación con miastenia gravis, enfermedad inflamatoria intestinal y hepatitis autoinmune, al tiempo que recientemente se ha relacionado su recurrencia como heraldo a algunos procesos neoplásicos de tipo hematolinfoide (síndrome mielodisplásico, sobre todo). Medicamentos como la penicilina y las tetraciclinas se han reportado como agentes disparadores en el desarrollo de la PANc, con mejoría clínica posterior a su suspensión inmediata17,18.
Por otro lado, está demostrada la presencia de anticuerpos IgM contra el complejo de protrombina-fosfatidil serina (anti-PS/PT) hasta en el 81,3% de los pacientes con PANc, con correlación positiva entre los niveles anti-PS/PT y proteína C reactiva (PCR)2,3, lo cual soporta la teoría de la unión de protrombina a las células apoptóticas endoteliales, desencadenando una respuesta inmunológica mediada por linfocitos T y B reactivos5,10,19. Además, existe activación de la vía clásica del complemento en PANc, al parecer secundaria al depósito de inmunoglobulinas tipo IgM e IgG.
Las manifestaciones cutáneas de la PAN clásica y de la PANc pueden ser muy similares. La PAN clásica involucra nódulos subcutáneos dolorosos en piernas (97% de los casos), cabeza y cuello (39%), brazos (33%) y tronco (8%)7,19. En un registro retrospectivo de 79 pacientes con PANc publicado por Daoud et al.20 se reportó que el 80% tenía nódulos dolorosos en las extremidades inferiores con edema; el 56% presentaba livedo reticular, el 49% úlceras y el 10% placas induradas. Además, se describió que hasta en el 31,3% de los pacientes había evidencia de lesiones hemorrágicas, desde petequias hasta púrpura extensa. No hay evidencia a la fecha de perforación septal anterior como manifestación clínica asociada de PANc, tal como se describió en el caso 1, en el que se descartaron las causas más frecuentes.
Nakamura et al.21 reportaron 22 pacientes en los que se encontraron nódulos subcutáneos en los miembros inferiores (100%) y los miembros superiores (16%); livedo reticular en los miembros inferiores (80%), el antebrazo (10%) y el dorso (10%); púrpura en los miembros inferiores (100%) y úlceras en los miembros inferiores (100%). Los nódulos estuvieron presentes en los 3 casos descritos y 2 de ellos presentaban livedo racemosa alrededor de una úlcera, lo que demuestra la variabilidad de localización y morfología de las lesiones. Lesiones de atrofia blanca, al igual que en el caso 3, se han reportado hasta en un 27,3%, y el dolor referido en la zona de los nódulos y las úlceras suele ser intenso17,21.
Los 3casos descritos aquí referían importante dolor de tipo lancinante precedido de mialgias, principalmente en el caso 1, lo que llevó a pensar en una afectación neuropática en el contexto de lepra22 y eritema nudoso leproso, o incluso en el curso de otras vasculitis asociadas con ANCA, como granulomatosis con poliangitis o poliangitis microscópica. La ausencia de compromiso visceral es lo que permite diferenciar la PANc de la variedad clásica, por lo que ante lesiones cutáneas características siempre se debe descartar el compromiso sistémico.
De igual forma, se deben tener presentes las manifestaciones extracutáneas que podrían acompañar a la PANc, tales como fiebre (18%), pérdida de peso (5%), neuropatía periférica (32%), mialgias (27%) y artralgias (18%), cuya presencia permitiría disminuir el umbral diagnóstico e intervenir el curso de la enfermedad de una manera más oportuna6,7,19.
No existen hasta la fecha pruebas diagnósticas específicas para la PANc. El diagnóstico se define por una correlación integral entre la clínica, los hallazgos histopatológicos y los laboratorios complementarios para descartar otras entidades. La tabla 2 describe una propuesta de diagnóstico clínico acogida por el Ministerio de Salud, Trabajo y Bienestar de Japón a partir del estudio de 22 casos de PANc21.
Criterios diagnósticos propuestos para PAN cutánea (PANc)
| Manifestaciones cutáneas | Nódulos subcutáneos, livedo reticular, púrpura, úlceras |
| Hallazgos histopatológicos | Vasculitis necrosante fibrinoide de arterias de pequeño y mediano calibre |
| Criterios de exclusióna | (1) Fiebre (= 38° C,=2 semanas), pérdida de peso (6kg o más en 6 meses)(2) Hipertensión arterial(3) Insuficiencia renal rápidamente progresiva, infarto renal(4) Hemorragia cerebral, infarto cerebral(5) Infarto de miocardio, cardiopatía isquémica, pericarditis, insuficiencia cardiaca(6) Pleuritis(7) Hemorragia intestinal, infarto intestinal(8) Neuropatía periférica fuera de la lesión cutánea afectada(9) Artralgia (artritis) o mialgia (miositis) fuera de la lesión cutánea(10) Arteriografía anormal (microaneurisma múltiple, estenosis y obliteración) |
| Decisión | Correlación entre hallazgos clínicos e histopatológicos y ausencia de todos los criterios de exclusión |
La PANc puede acompañarse de neuropatía periférica y mialgia, siempre y cuando estos síntomas se limiten a la misma área ocupada por lesiones cutáneas.
Tomado de Nakamura et al.21.
Las series japonesas, europeas y turcas sugieren realizar como estudios iniciales hemograma completo, eritrosedimentación, PCR, función renal y bioquímica hepática completa, además de descartar infección por VIH, VHB y VHC23-25. Usualmente, las pruebas serológicas para sífilis, ANA, anti-ENA, ANCA, FR y anti-CCP son negativas; sin embargo, se ha descrito positividad para p-ANCAS en el grupo de pacientes con PANc inducida por minociclina15,23. Incluso, hasta el 43,8% puede tener anticoagulante lúpico positivo a títulos variables3,4.
La histopatología se caracteriza por la presencia de vasculitis leucocitoclástica y degeneración fibrinoide de pequeños y medianos vasos de la dermis y el tejido celular subcutáneo7,18. Se han descrito 4 estadios que muestran la evolución de la PANc en la histopatología: el primero o degenerativo, caracterizado por depósito de material fibrinoide y destrucción de la lámina elástica; el segundo o inflamatorio, con infiltrado neutrofílico y algunos eosinófilos de distribución perivascular; el tercero o de granulación, conformado por infiltrado linfohistiocitario, con proliferación de la íntima y trombosis de la arteria; y el cuarto o estadio final, con proliferación fibroblástica perivascular1-3,21.
Gupta et al.24 describieron la inflamación del panículo adiposo en PANc, con un infiltrado neutrofílico y algunos eosinófilos perivasculares localizados en el lobulillo graso, lo cual era causa de isquemia y ulceración. Los 3 casos presentaron vasculitis de pequeño y mediano vaso, además de paniculitis, tal como se describió en la paciente del caso 1.
En cuanto a la inmunofluorescencia directa, se podría observar depósito perivascular de IgM o complemento, lo cual apoya la teoría de los inmunocomplejos. En el caso 2 se observó adicionalmente depósito de C1q, lo cual obligaba a descartar diagnósticos diferenciales como el lupus eritematoso sistémico dado que estos hallazgos son propios de la denominada banda lúpica.
Las exacerbaciones son frecuentes y pueden durar entre 2 y 8 semanas. Como factores asociados con peor pronóstico (recurrencia en los siguientes 2 años de seguimiento) se han descrito los siguientes: múltiples úlceras en el inicio y reactantes de fase aguda elevados antes de iniciar el tratamiento25-27.
Por su parte, Chen28 reportó que los pacientes con mialgias, artralgias y síntomas asociados con compromiso de nervio periférico (disestesias, parestesias) tenían recaídas más frecuentes y severas26. La progresión de PANc a clásica se ha reportado hasta en el 15-20% de los pacientes, por lo que se debe descartar progresión durante cada recaída8-10. Se ha descrito mayor progresión sistémica en pacientes con FR y ANA positivos, a títulos altos29-31.
Hasta el momento, no hay estudios prospectivos ni guías basadas en evidencia para el tratamiento de la PANc. Las estrategias terapéuticas se basan en reportes de caso, series de caso y revisiones de la literatura. La tabla 3 resume los tratamientos y las dosis que se describen en la literatura a la fecha. El tratamiento depende de las manifestaciones clínicas y la severidad29.
Dosis y esquemas terapéuticos de PAN cutánea (PANc)
| Severidad | |||
|---|---|---|---|
| Opciónterapéutica | Leve | Severa | Refractaria |
| Primera línea | Furoato de mometasona 0,1% crema, propionato de clobetasol 0,05% crema o ungüento: 2 veces al día sobre la lesión en oclusión por 8-12 semanas27,31,34AINE: ibuprofeno hasta 800mg cada 8 h, naproxeno 500mg cada 12 h27,30Colchicina: 0,5-1 mg/día30 | Prednisolona: 30 mg/día-1 mg/kg/día durante al menos 4 semanas y realizar descenso progresivo hasta dosis efectiva mínima21,30,31Azatioprina: 2-3 mg/kg/día30,37Metotrexato: 15-22,5 mg/semanales31,36Cloroquina: 3 mg/kg/día31,38Hidroxicloroquina: 6 mg/kg/día31,38Micofenolato mofetilo: 1-3 g/día35 | Inmunoglobulina intravenosa: 1-2 g/kg cada 4 semanas41,42Infliximab: 5 mg/kg semana 0, 2643,46Etanercept: 50-100 mg/semanal44-46 |
En la PANc leve se indican como primera línea el reposo relativo, los cambios de posición periódicos (incluyendo posición de Trendelenburg), antiinflamatorios no esteroideos (AINE) o colchicina; en caso de lesiones muy localizadas o limitadas, se pueden añadir esteroides tópicos de alta potencia, como clobetasol31-34. El uso de dapsona ha sido reportado con resultados variables32. La pentoxifilina y la sulfasalazina podrían ser usadas como terapia adyuvante, nunca como monoterapia31,34.
En pacientes con compromiso más extenso o en casos severos, la prednisolona por vía oral es el tratamiento de elección30,32,33. En los 3casos presentados, esta fue la terapia de elección, a las dosis recomendadas por la literatura. El uso de azatioprina, metotrexato, antimaláricos (cloroquina, hidroxicloroquina) y micofenolato de mofetilo es frecuentemente requerido como terapia inmunosupresora adyuvante35,36, dado que estos medicamentos permiten desescalonamiento en dosis de esteroide, mejor control de los síntomas y disminución en las recaídas4,31-34,37.
En pacientes con mal control de la enfermedad, el uso de ciclofosfamida IV en pulsos, asociado con el esteroide por vía oral, ha mostrado altas tasas de remisión. En estos casos, se reseña el esquema habitualmente indicado para vasculitis asociadas con ANCA en fase de inducción: 0,75mg×m2 de área de superficie corporal IV cada 4 semanas, sin superar 1g por infusión por 6 meses. En pacientes con deterioro de la función renal se modifica la dosis así: 0,5mg×m2 de área de superficie corporal; de igual forma, en pacientes mayores de 65 años la dosis por pulso será de 500mg IV cada 15 días, para un total de 6 dosis38-40.
La inmunoglobulina IV es la última línea terapéutica en casos de enfermedad refractaria41,42, al igual que agentes dirigidos contra el factor de necrosis tumoral alfa (TNF-α), particularmente infliximab o etanercept43,44. Otras opciones terapéuticas, como tamoxifeno, oxígeno hiperbárico, warfarina y factor estimulante de colonias de granulocitos (G-CSF, en inglés), también se han reportado como coadyuvantes, con evidencia clínica limitada a la fecha45-50.
El caso 1 demuestra no solo la importancia de hacer un adecuado enfoque diagnóstico, a partir de la exclusión de los diagnósticos más frecuentes asociados con las lesiones descritas, pasando por el compromiso multisistémico propio de la PAN clásica hasta llegar a manifestaciones cutáneas circunscritas, sino también las implicaciones terapéuticas y pronósticas que implican casos de alta recurrencia o aparición de nuevas lesiones, incluyendo aun terapia biológica dentro de las alternativas de manejo.
Con respecto a este mismo caso, se presentan varias dificultades en términos diagnósticos y de seguimiento:
El cuadro clínico inició con nódulos subcutáneos difusos, asociado con perforación del tabique nasal; estuvo con inmunosupresión de forma previa, lo que puede enmascarar la presentación actual de las manifestaciones documentadas.
No se cuenta con biopsia renal ni angio-TAC o arteriografía que descarten la afectación de vasos abdominales de mediano calibre, por lo que no es posible descartar del todo vasculitis de pequeños vasos o PAN sistémica.
La respuesta a infliximab puede observarse en PAN sistémica.
ConclusionesLa PANc es una variante clínica poco frecuente dentro del espectro de vasculitis de medianos y pequeños vasos. Se caracteriza por la presencia de nódulos subcutáneos dolorosos en las piernas, asociados con livedo reticular, con alto riesgo de ulceración. Tiene un curso benigno, crónico y variable, con altas tasas de recurrencia y asociación con procesos tanto infecciosos (VIH, virus hepatotropos, tuberculosis, lepra) como no infecciosos (medicamentos, enfermedades del tejido conectivo); en ocasiones se considera fenómeno paraneoplásico principalmente de neoplasias hematolinfoides.
El diagnóstico de la PANc es desafiante, ya que no existe un marcador específico de la enfermedad y las manifestaciones clínicas pueden solaparse con otras entidades inflamatorias, infecciosas e incluso relacionadas con fenómenos de hipersensibilidad a algunos fármacos. Según las características de las lesiones y los síntomas relacionados, entre los que se destacan las mialgias, la indicación temprana de biopsia de piel es primordial para cercar la lista de diagnósticos posibles y llegar a la PANc, tras un proceso de razonamiento clínico de exclusión.
Hasta la fecha, no existen guías de tratamiento para PANc. Se recomienda iniciar con esteroides sistémicos, asociados con otros inmunomoduladores que permitan un desmonte temprano de las dosis convencionales. De igual forma, hay reportes anecdóticos de respuesta a citostáticos en esquemas de inducción propuestos para pacientes con vasculitis asociadas con ANCA, así como la administración de terapia biológica (agentes anti-TNF-α, como en el caso 1) en pacientes con predictores de mal pronóstico, incluyendo inicio con múltiples lesiones y alta tasa de recurrencia en el primer año.
LimitacionesEsta serie de 3casos cuenta con algunas limitaciones. A causa de la pandemia por COVID-19 se ha perdido el seguimiento de los pacientes, tanto presencial como remotamente, sin que a la fecha se conozca su evolución clínica. Así mismo, para algunos medicamentos inmunomoduladores no se logró documentar la dosis exacta (como en el caso 1) por ser indicados extrainstitucionalmente y no contar con historia clínica previa.
Responsabilidades éticasProtección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni animales.
Confidencialidad de los datos. Los autores declaran que han seguido los protocolos del centro hospitalario de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los autores han obtenido el consentimiento informado de los pacientes referidos en el artículo.
FinanciaciónEste trabajo no recibió financiación de ningún tipo.
Conflicto de interesesLos autores declaran no tener conflicto de intereses.
