



The combination of sarcoidosis and lymphoma (sarcoidosis-lymphoma syndrome) is a rare but recognized clinical condition. Some manifestations may be common among these conditions, so differentiating each individual disease is challenging for the clinician, and even more so if they coexist in one same patient. In this latter scenario, neither the clinical manifestations, nor the laboratory parameters, are specific; therefore, it is mandatory to do a meticulous analysis of each case, supported by laboratory tests, imaging studies, and histopathology in order to arrive at an accurate diagnosis. This article discusses two cases of sarcoidosis-lymphoma syndrome, with an analysis of the key aspects in the diagnosis of this clinical condition.
La combinación de sarcoidosis y linfoma (Síndrome sarcoidosis-linfoma) es una asociación poco frecuente pero reconocida en el ámbito clínico. Algunas manifestaciones pueden ser comunes entre estas entidades, por lo que es un reto para el clínico diferenciar cada enfermedad por separado o si coexisten en un mismo individuo. En este último escenario, ni las manifestaciones clínicas ni los parámetros de laboratorio son específicos, por lo que es necesario un análisis minucioso de cada caso, apoyado de laboratorios, imágenes e histopatología para llegar a un diagnóstico certero. Se presentan dos casos de síndrome sarcoidosis-linfoma y se analizan los aspectos clave en el diagnóstico de esta forma clínica.
The association of sarcoidosis with malignant processes was initially described by Brincker and Wilbek1 in 1974, and later in 1986 Brincker2 defined the sarcoidosis-lymphoma syndrome and described its major characteristics: 1) the lymphoproliferative process is usually diagnosed after sarcoidosis; 2) the mean age at diagnosis of sarcoidosis is 41 years, 10 years older than the usual cases of sarcoidosis; and 3) Hodgkin disease is more frequent among patients with sarcoidosis than in the general population.3,4 It is believed that its pathophysiological mechanisms may be associated with a dysfunction of the cell immune system that causes the clonal proliferation of B lymphocytes.4 However, there is some controversy when considering probable misdiagnoses of sarcoidosis, particularly when only cytological techniques are used, since any lymphomas associated with an extensive granulomatous response may blur the diagnosis of malignancy.
It has been said that the development of adenopathies and splenic dysfunction during a chronic presentation of sarcoidosis should be investigated early, in order to rule out the concomitant development of lymphoma, since the development of adenopathies occurs in over 60% of lymphomas, while splenomegaly develops in 30–70% of the cases; however, in patients with sarcoidosis, splenomegaly is only present in 7% of the cases.5
Clinical case 1This is a 21-year old female patient, born in Montería and resident in Medellín, studying to become a flight attendant, single mother with one daughter, with no relevant medical history. She came to the doctor after 8 months of evolution of a clinical condition involving objective weight loss (9kg), associated with subjective fever, dry cough, paresthesia of the lower extremities and fatigue. The initial findings were anemia and cervical adenomegalies; a chest CT was performed resulting in bilateral hilar adenomegalies. The patient was hospitalized. Multiple studies were conducted while the patient was hospitalized, including bone marrow aspirate and biopsy, infectious, autoimmune and neoplastic disease were ruled out. Later, a mediastinum endoscopy was performed with a mediastinal lymph bone biopsy resulting in liquefaction necrosis, but no complementary studies of the specimen were performed (special stains, immunohistochemistry, etc.). Four months after the onset of the clinical manifestations, the patient developed painful purplish nodes in the anterior aspect of both legs, associated with ankle edema.
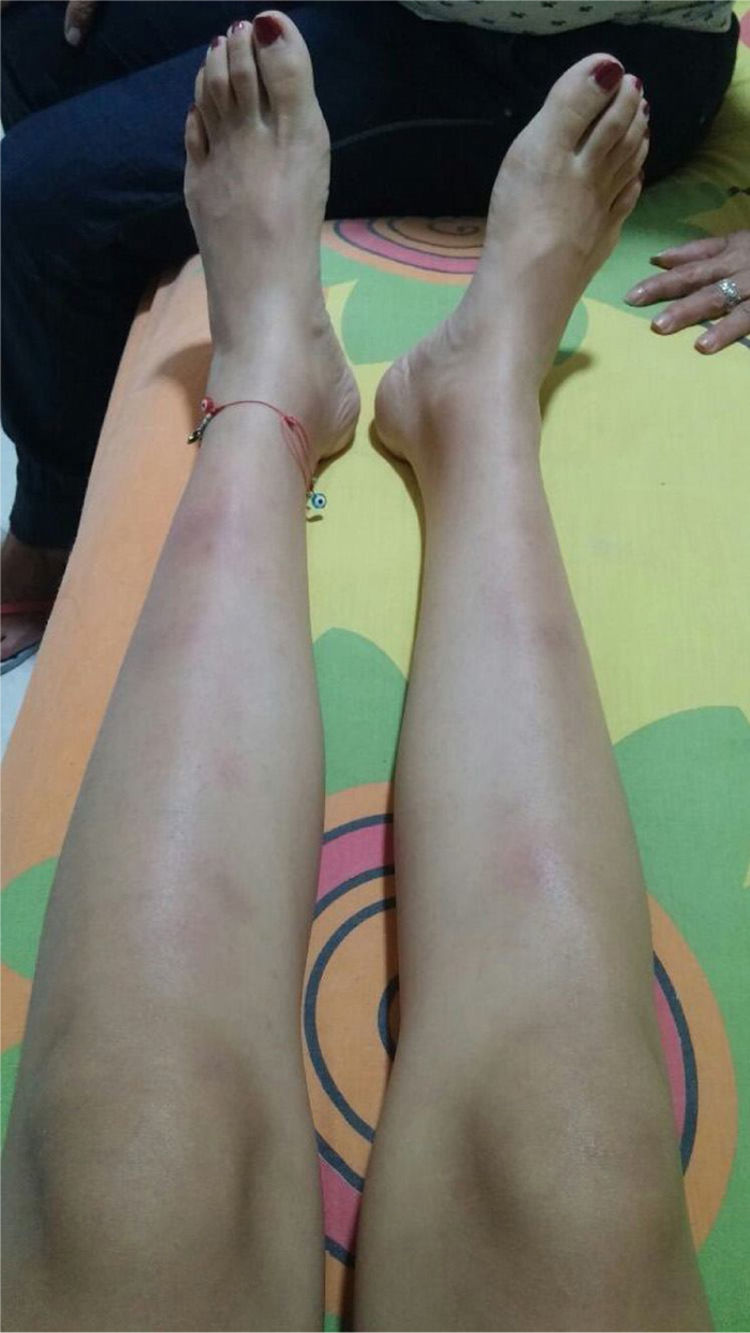
The symptoms deteriorated and the patient was admitted to our institution. At admission, the patient was normotensive, tachycardic, afebrile, with pale conjunctivas and multiple clusters of cervical lymph nodes, painful erythematous purplish nodes in the anterior aspect of both extremities (Fig. 1). The laboratory findings showed mild anemia, elevated acute phase reactants, normal liver function tests, preserved kidney function and negative antibodies (Table 1). The bronchoalveolar lavage was normal, with negative microbiological studies (bacteria, mycobacteria and fungi). Cervical CT with multiple adenopathies in the carotid space and in the right posterior cervical space of the neck, with a short axis of 17.5 and 14mm, and towards the supraclavicular region. The chest CT revealed adenopathies in the anterior and medial mediastinum, of pre-vascular, pre-tracheal, retrocaval localization with a diameter of 44×37mm, right paratracheal, subcarinal, and towards both hilar regions. The abdominal CT was unremarkable. The patient was considered highly suspicious of sarcoidosis since she met the Löfgren syndrome criteria (bilateral hilar adenopathy, knee arthritis and erythema nodosum), which has a 95% specificity for the diagnosis of acute sarcoidosis. A new excision biopsy of the cervical lymph node was conducted and the result was large strongly positive Reed-Sternberg/Hodgkin CD30 cells, weakly positive CD20, weakly positive PAX-5, negative BOB1, negative BCL6, negative BCL2, negative CD10, positive CD23, negative EMA, negative latent membrane protein 1 (LMP-1), negative CD3, with abundant surrounding T lymphocytes, in cycle with KI67. The reticulin stain shows increased fiber pattern with sclerotic appearance delimiting the lymph nodes. Conclusion: classical Hodgkin lymphoma, nodular sclerosis type grade 1, partial CD20 expression. The patient was assessed by hematology and high-risk polychemotherapy was initiated with Adriamycin, vinblastine, bleomycin, dacarbazine (ABVD) achieving a good response and the patient is currently in remission of the disease.

Laboratory findings.
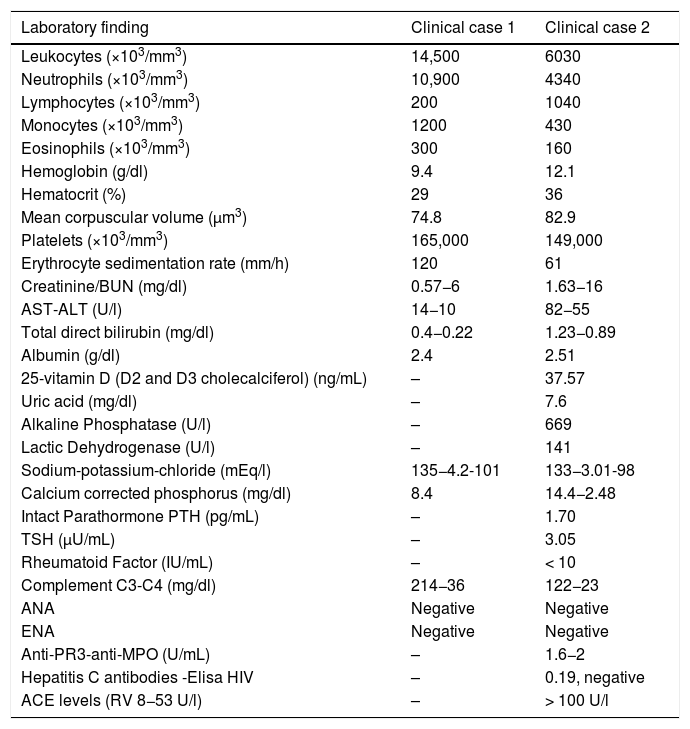
| Laboratory finding | Clinical case 1 | Clinical case 2 |
|---|---|---|
| Leukocytes (×103/mm3) | 14,500 | 6030 |
| Neutrophils (×103/mm3) | 10,900 | 4340 |
| Lymphocytes (×103/mm3) | 200 | 1040 |
| Monocytes (×103/mm3) | 1200 | 430 |
| Eosinophils (×103/mm3) | 300 | 160 |
| Hemoglobin (g/dl) | 9.4 | 12.1 |
| Hematocrit (%) | 29 | 36 |
| Mean corpuscular volume (μm3) | 74.8 | 82.9 |
| Platelets (×103/mm3) | 165,000 | 149,000 |
| Erythrocyte sedimentation rate (mm/h) | 120 | 61 |
| Creatinine/BUN (mg/dl) | 0.57−6 | 1.63−16 |
| AST-ALT (U/l) | 14−10 | 82−55 |
| Total direct bilirubin (mg/dl) | 0.4−0.22 | 1.23−0.89 |
| Albumin (g/dl) | 2.4 | 2.51 |
| 25-vitamin D (D2 and D3 cholecalciferol) (ng/mL) | – | 37.57 |
| Uric acid (mg/dl) | – | 7.6 |
| Alkaline Phosphatase (U/l) | – | 669 |
| Lactic Dehydrogenase (U/l) | – | 141 |
| Sodium-potassium-chloride (mEq/l) | 135−4.2-101 | 133−3.01-98 |
| Calcium corrected phosphorus (mg/dl) | 8.4 | 14.4−2.48 |
| Intact Parathormone PTH (pg/mL) | – | 1.70 |
| TSH (μU/mL) | – | 3.05 |
| Rheumatoid Factor (IU/mL) | – | < 10 |
| Complement C3-C4 (mg/dl) | 214−36 | 122−23 |
| ANA | Negative | Negative |
| ENA | Negative | Negative |
| Anti-PR3-anti-MPO (U/mL) | – | 1.6−2 |
| Hepatitis C antibodies -Elisa HIV | – | 0.19, negative |
| ACE levels (RV 8−53 U/l) | – | > 100 U/l |
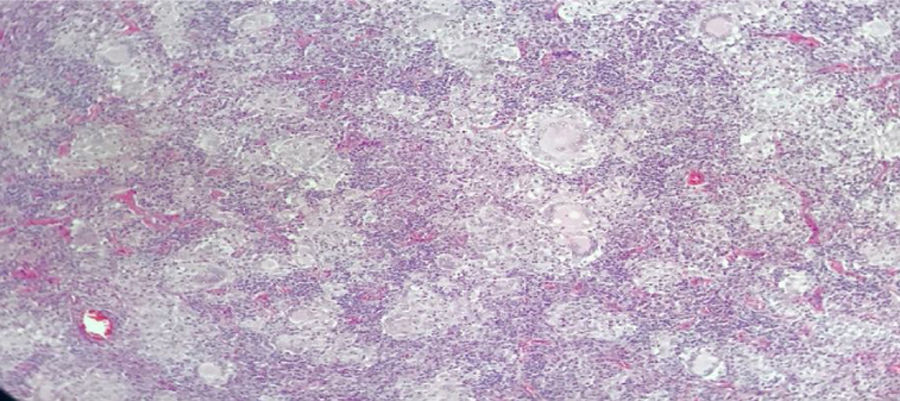
Male, 43-year old patient, mestizo race, farmer, with no relevant personal or family history. In 2016 began to experience generalized malaise, asthenia, adynamy, febriculas, symmetrical polyarthralgias in the hands (particularly in the proximal interphalangeal joints), wrists, and knees, which lasted throughout the day, with no morning stiffness, associated with non-painful cervical adenopathies. A cervical lymph node biopsy was performed in December 2016, reporting: chronic granulomatous inflammation without necrosis (Fig. 2). The symptoms deteriorated in March 2017 and the patient was referred for a level 4 institution, and was admitted to our institution. The initial physical examination revealed a patient with a chronic illness, with body weight of 61kg (his usual weight was 68kg), with bilateral anterior and posterior cervical chain adenopathies of approximately 1−2cm, not painful; in the abdomen the liver was palpable 1cm below the costal margin, not painful; additionally there were inguinal adenopathies of approximately 1cm, not painful; no joint deformity and no synovitis was identified. The neck CT showed lymph node clusters on both sides of the neck; the chest CT showed enlarged lymph nodes of the mediastinum and in the axillary regions, alveolar opacities converging in the anterior and posterior segment of the upper left lobe, associated with ground glass opacities in the posterior segments of both lung fields. Visible lytic lesions in the vertebral bodies of T4, T5, T7 and T8; the abdominal CT revealed retroperitoneal lymph node clustering and hepatosplenomegaly.

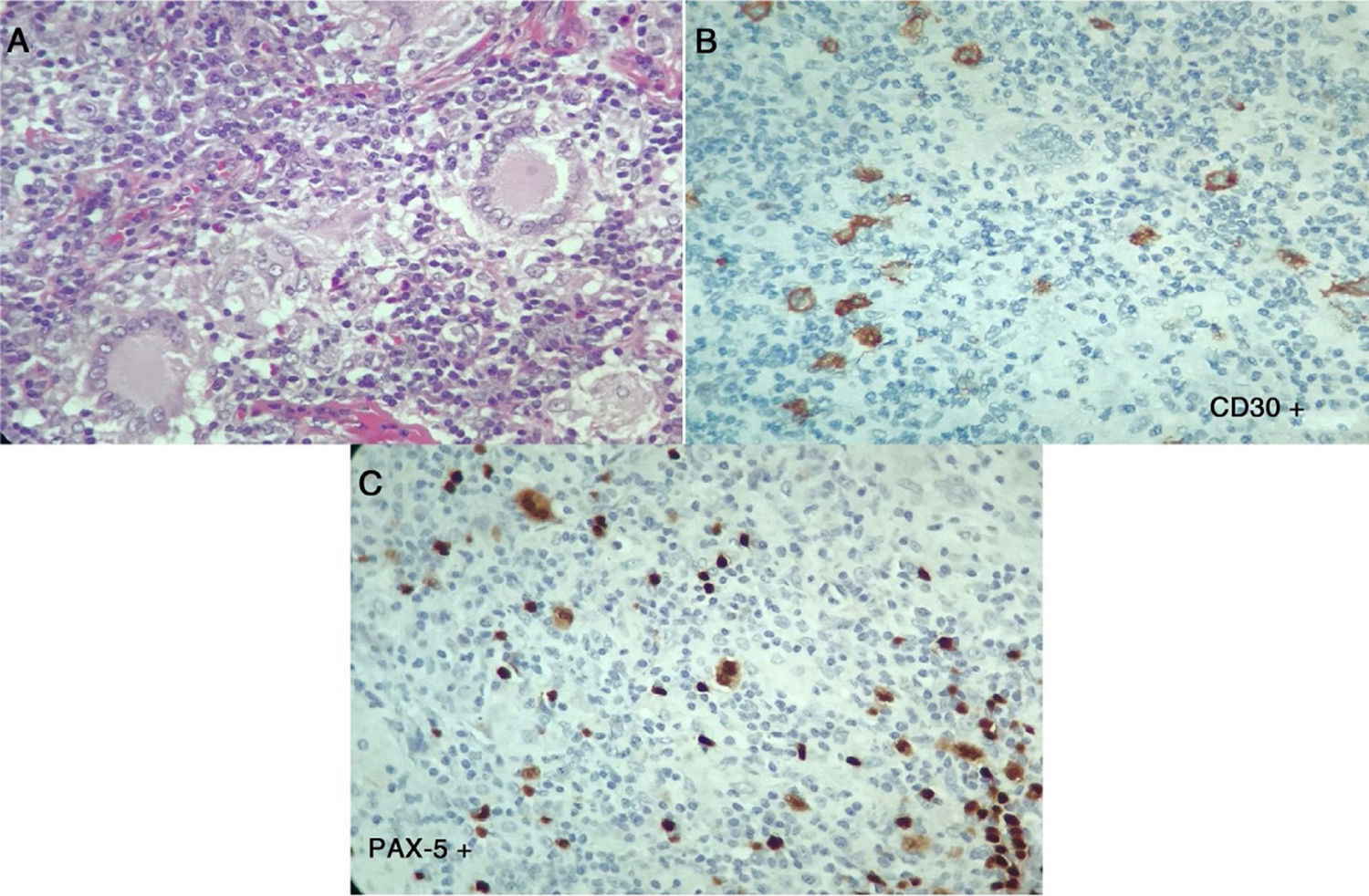
The laboratory tests (Table 1), reported normal blood count, with elevated acute phase reactants, azotemia without creatinine/urea nitrogen dissociation, mild transaminase elevation, hypoalbuminemia, elevated alkaline phosphatase, hypercalcemia with suppressed PTH, normal levels of 25-vitamin D, occasional high urinary calcium, negative autoimmunity panel and extremely high levels of angiotensin converting enzyme (ACE), all leading to a diagnosis of sarcoidosis. The patient was admitted to the Intensive Care Unit (ICU) for hypercalcemia management. In view of a potential diagnosis of an associated neoplasm, the cervical lymph node biopsy was repeated on April 7, 2017; additionally, there were dorsal spine lesions reported with multinucleated giant cell lymph node expressing CD30 – strongly positive -, MUM-1, PAX-5 and LMP-1. The lymphoid population is mostly T-helper cells CD3, CD4, CD2, CD7, CD5, CD25 with few CD8. These results are compatible with classical Hodgkin lymphoma, nodular sclerosis with spinal infiltration, associated with non-caseating granulomatous lesions (Fig. 3). The patient was diagnosed with sarcoidosis-lymphoma syndrome and the ABVD protocol was initiated on April 9, 2017, in addition to prednisolone 5mg per day. The patient responded favorable until this date.
 Lymph node with abnormal architecture, large, lobulated, binucleated and mononucleated cells, with prominent nucleolus, multinucleated giant cells. Additionally, fibrosis and stroma collagen formation are present. B and C) The neoplastic cells described express CD30, CD15, MUM-1, PAX-5 and latent membrane protein 1 (LMP-1). The lymphoid population is mostly helper T-cells CD3, CD4, CD2, CD7, CD5, with few CD8. Compatible with classical Hodgkin lymphoma, nodular sclerosis.")
Second histopathology of the cervical lymph node, Case 2. A) Lymph node with abnormal architecture, large, lobulated, binucleated and mononucleated cells, with prominent nucleolus, multinucleated giant cells. Additionally, fibrosis and stroma collagen formation are present. B and C) The neoplastic cells described express CD30, CD15, MUM-1, PAX-5 and latent membrane protein 1 (LMP-1). The lymphoid population is mostly helper T-cells CD3, CD4, CD2, CD7, CD5, with few CD8. Compatible with classical Hodgkin lymphoma, nodular sclerosis.
Sarcoidosis is an inflammatory disease of unknown etiology, with a broad spectrum of presentations requiring a compatible clinical – radiological assessment and a histology report of epithelioid non-caseating granulomas.5,6 The manifestations of the disease may be quite varied, but the most frequent are fatigability, persistent coughing, and skin, ocular, lymph node and lung involvement; the former is present in up to 90% of the cases.6 With regards to the skin manifestations, the erythema nodosum should be highlighted, which is part of Löfgren syndrome, corresponding to one of the acute forms of sarcoidosis, characterized by hilar adenopathies, arthritis, fever and erythema nodosum, as described in Case 1. The final diagnosis of the disease requires the histological confirmation of non-caseating epithelioid granulomas, having previously ruled out other causes that could account for these findings, such as infectious diseases (tuberculosis, leishmaniasis, toxoplasmosis, mycosis, brucellosis, inter alia) or lymphoproliferative conditions (Hodgkin lymphoma).
There is an association between sarcoidosis and malignancy that may present in different forms. Patients with sarcoidosis may develop malignancies, just as cancer patients may develop sarcoid reactions. Sarcoid reactions are considered when epithelioid non-caseating granulomas develop in patients who do not meet the criteria for systemic sarcoidosis. Granulomas with sarcoid-like reactions may present associated with Hodgkin (96.4%) or Non-Hodgkin (3.6%) lymphoma, and may be so extensive than mask the diagnosis of a malignant process.4 The association between sarcoidosis-lymphoma was described for the first time in a group of 46 cases in which there was a relationship between sarcoidosis and the development of lymphoproliferative disease.2 The sarcoidosis-lymphoma syndrome refers classically to the development of lymphoma at least 1–2 years after the diagnosis of sarcoidosis4; however, there are case reports with less than one-year difference.7–9
The non-caseating granulomas observed in sarcoidosis may be detected concomitantly in the malignant lymphoproliferative disease, even when the clinical manifestations do present even several months later. It has been estimated that the mean interval between the development of sarcoidosis and lymphoma es 24 months, but there have been cases occurring decades later. Middle-aged individuals presenting with chronic active sarcoidosis, have a 5-fold incidence of lymphoproliferative diseases and fifty percent of the cases these are pulmonary low-grade lymphomas.10 A Swedish cohort of patients with sarcoidosis showed a relative high risk for non-Hodgkin lymphoma, leukemia, and lung, stomach, small bowel, liver and skin cancer. Chronic inflammation is suggested as the presumptive mediator for increased risk of cancer, particularly in the organs affected by the sarcoidosis.3 Persistent sarcoidosis activity, anergy, and lymphopenia are associated with the syndrome.11 It has been suggested that the etiology of cancer-related sarcoidosis is secondary to a T-cell mediated host response, induced by soluble antigenic tumor factors. The antigenic factors may be eliminated by the tumor cells or released during the tumor necrosis. Then, the lymph vessels transport the factors to the lymph nodes, where the host immune response – similar to a type IV hypersensitivity reaction – results in the development of non-caseating epithelioid granulomas. Moreover, there is hyperactivity of the B cells system leading to increased mitotic lymphocyte activity which increases the risk of mutation and malignant transformation.4 Both in sarcoidosis and in lymphoma, the role of the Epstein-Barr virus is considered the cause of genetic transformation, leading to the development of the disease.12 Hypercalcemia and increased ACE are associated with sarcoidosis, but there have been also cases of patients with an isolated lymphoma. Sarcoidosis is diagnosed based on clinical and histological criteria, upon ruling out the presence of a lymphoproliferative disease, through an anatomopathological study of an adenopathy, defining its morphological characteristics, assisted by immunophenotyping techniques, using specific markers such as CD20, CD3, bcl2, CD5, inter alia.
There is no specific therapy for this condition; the treatment will depend on the type of neoplasm, while the mainstay of therapy for sarcoidosis is the use of steroids.13
ConclusionSarcoidosis and some lymphomas do not only share several clinical manifestations, but also analytical and even histological findings. In the presence of multiple adenopathies, an underlying lymphoproliferative process should be ruled out through an anatomopathological study and immunohistochemistry techniques, even in the presence of a sarcoidosis-based diagnosis, since both entities may coexist, particularly if there is a previous diagnosis of sarcoidosis and the disease experienced a clinical change over time, which allowed for the identification of an underlying lymphoma.
Conflict of interestsThe authors have no conflict of interest to disclose.
Please cite this article as: Aguirre-Valencia D, Maturana JV, Tabares SL, Bravo J-C, Tobón GJ, Vásquez GM. Síndrome sarcoidosis-linfoma. Presentación de dos casos clínicos. Rev Colomb Reumatol. 2020;27:298–302.
