




Splenic infarction is a rare condition usually associated with systemic conditions. Antiphospholipid syndrome (APS) is an autoimmune condition that can present as abdominal pain due to splenic infarction. We describe a patient with systemic lupus erythematosus, admitted due to acute abdominal pain. Abdominal CT scan showed splenic infarction, and tests for antiphospholipid and anti-β2-glycoprotein I antibodies were positive. The pathophysiology and clinical presentation of splenic infarction associated with APS is discussed, emphasizing the importance of including this entity in the differential diagnosis of abdominal pain of uncertain cause in patients with systemic conditions.
El infarto esplénico es una condición infrecuente, generalmente asociada con afecciones sistémicas. El síndrome antifosfolipídico (SAF) es una enfermedad autoinmune que puede presentarse como dolor abdominal debido a infarto esplénico. Se describe el caso de una paciente con lupus eritematoso sistémico que ingresó con dolor abdominal agudo. La tomografía abdominal evidenció infarto esplénico y las pruebas de laboratorio para anticuerpos antifosfolípidos y anti-β2-glicoproteína I fueron positivas. Se discute la fisiopatología y la presentación clínica del infarto esplénico asociado con SAF, enfatizando la importancia de incluir esta entidad en el diagnóstico diferencial del dolor abdominal de causa incierta en pacientes con afecciones sistémicas.
Splenic infarction is a relatively uncommon condition in clinical practice. There are various causes, among which the most common are thromboembolic diseases, such as those associated with atrial fibrillation, cardiac procedures, bacterial vegetations generated in the heart valves, or related to valvular disease.1
Non-embolic etiologies of splenic infarction remain rare, including some autoimmune diseases (systemic lupus erythematosus -SLE-, vasculitis), hypercoagulable states (e.g., antiphospholipid syndrome [APS]), or malignancies. Other rarely reported causes comprise hematological disorders such as polycythemia vera, myeloproliferative diseases, lymphomas, leukemias, as well as hemolytic anemias and hemoglobinopathies (particularly sickle cell anemia), and, among the infectious causes, bacterial endocarditis is described.1,2
Wand et al.3 reported the mechanisms underlying the development of splenic infarcts, among which cardioembolic infarcts represented 54.4%, vascular disorders 20%, and hematological diseases 15.6%. Atrial fibrillation and atherosclerosis were the most common disorders in patients over 70 years of age, while APS predominated in young people.
APS is a systemic autoimmune disorder that can cause thromboembolic events or obstetric complications associated with antiphospholipid antibodies. It can be divided into primary or secondary, depending on the presence of an underlying autoimmune disease, such as rheumatoid arthritis or SLE.4
We present the case of a 41-year-old patient, with a history of untreated SLE, who was admitted for abdominal pain, hemolytic anemia, jaundice, and hepatosplenomegaly, being diagnosed with active SLE related to APS and autoimmune hemolytic anemia.
Case presentationA 41-year-old woman, mestizo, originally from metropolitan Lima, with a history of SLE diagnosed 5 years ago due to malar erythema, alopecia, positive antinuclear antibodies, and hypocomplementemia. She was treated with prednisone at varying doses for the first 3 years, after which she discontinued due to lack of symptoms. Four years ago, during her third and final pregnancy, at 33 weeks, she developed severe preeclampsia, which resulted in a stillbirth.
The patient assisted to a low-level complexity hospital due to 6 days of diffuse, stabbing abdominal pain, of moderate intensity, predominantly in the upper abdomen. She was administered parenteral analgesics and was instructed to continue with outpatient management. Three days later, the abdominal pain reappeared, acutely exacerbated during the last 24 hours, which intensified with deep inspiration, associated with asthenia, progressive hyporexia, and nausea. A hemoglobin level of 3.5 g/dL was found. For all these reasons, it was decided to transfer her to the emergency service of the Hipólito Unánue National Hospital.
Upon admission, the patient persisted with abdominal pain, associated with bilious vomiting. On initial assessment, she was normotensive, tachycardic, with painful facies, and antalgic position. Marked jaundice was evident in scleras and mucous membranes; abdomen with air-fluid sounds, soft, depressible, without resistance in the abdominal wall, and painful on palpation in the left hypochondrium. Traube’s space was occupied, the spleen was palpated one centimeter below the costal margin, while the rest of the examination was unremarkable.
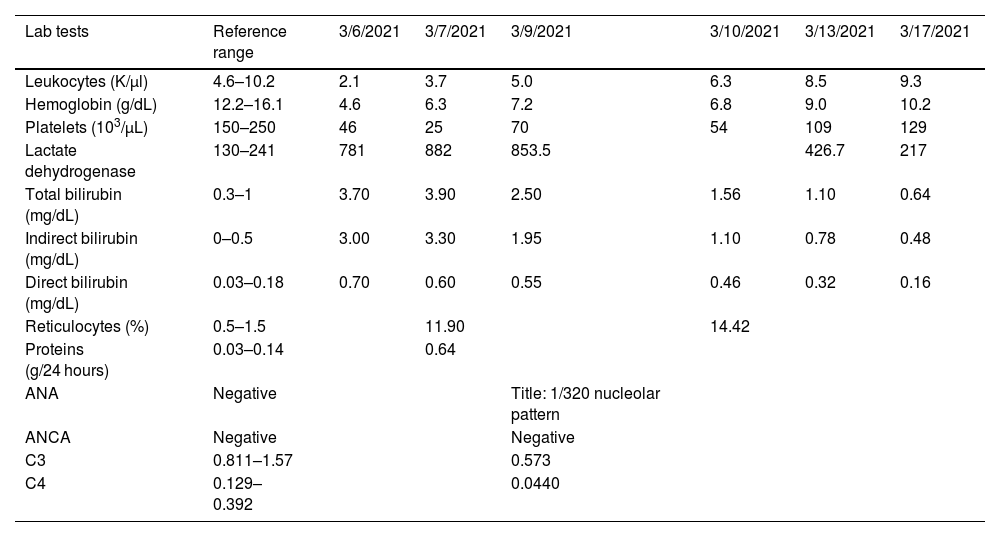
The patient underwent general blood tests (Table 1), which revealed the presence of severe anemia, leukopenia, severe thrombocytopenia, and predominantly indirect hyperbilirubinemia. The presence of elevated reticulocytes (corrected reticulocyte production index of 3.0), a positive direct Coombs test (+++ according to qualitative determination with 16 dilution titer), and elevated lactate dehydrogenase confirmed the diagnosis of hemolytic anemia. These findings, along with the presence of leukopenia, thrombocytopenia, positive anti-native DNA (22.1IU/ml), hypocomplementemia, and granular casts in urinalysis confirmed lupus activity, with an activity index (SLEDAI) of 10 points.
Blood analysis.
| Lab tests | Reference range | 3/6/2021 | 3/7/2021 | 3/9/2021 | 3/10/2021 | 3/13/2021 | 3/17/2021 |
|---|---|---|---|---|---|---|---|
| Leukocytes (K/µl) | 4.6–10.2 | 2.1 | 3.7 | 5.0 | 6.3 | 8.5 | 9.3 |
| Hemoglobin (g/dL) | 12.2–16.1 | 4.6 | 6.3 | 7.2 | 6.8 | 9.0 | 10.2 |
| Platelets (103/µL) | 150–250 | 46 | 25 | 70 | 54 | 109 | 129 |
| Lactate dehydrogenase | 130–241 | 781 | 882 | 853.5 | 426.7 | 217 | |
| Total bilirubin (mg/dL) | 0.3–1 | 3.70 | 3.90 | 2.50 | 1.56 | 1.10 | 0.64 |
| Indirect bilirubin (mg/dL) | 0–0.5 | 3.00 | 3.30 | 1.95 | 1.10 | 0.78 | 0.48 |
| Direct bilirubin (mg/dL) | 0.03–0.18 | 0.70 | 0.60 | 0.55 | 0.46 | 0.32 | 0.16 |
| Reticulocytes (%) | 0.5–1.5 | 11.90 | 14.42 | ||||
| Proteins (g/24 hours) | 0.03–0.14 | 0.64 | |||||
| ANA | Negative | Title: 1/320 nucleolar pattern | |||||
| ANCA | Negative | Negative | |||||
| C3 | 0.811–1.57 | 0.573 | |||||
| C4 | 0.129–0.392 | 0.0440 |
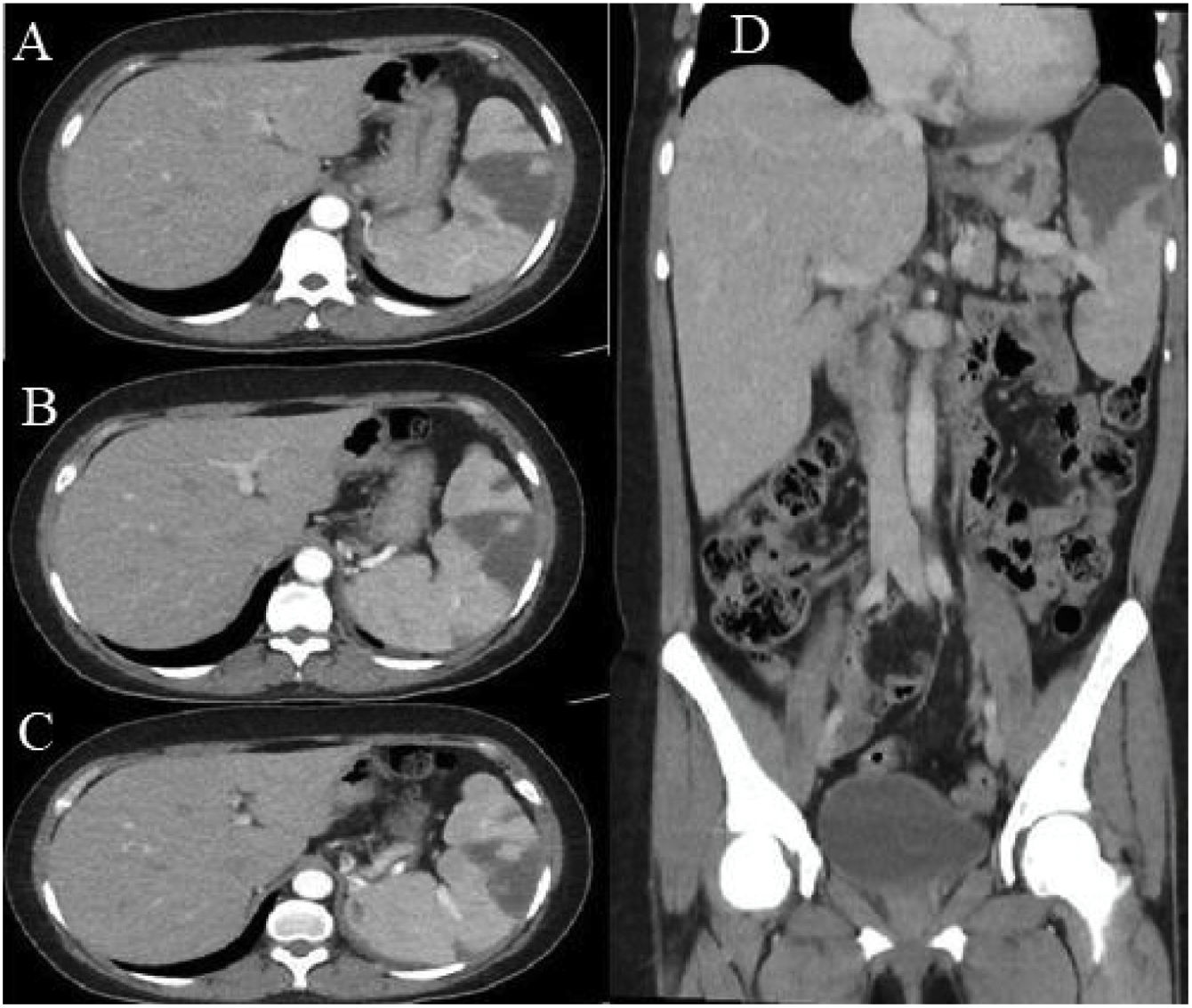
Abdominal ultrasound showed hepatosplenomegaly, with the right hepatic lobe measuring 174 mm, the left lobe measuring 105 mm, and the spleen measuring 147 × 84 mm, the latter with multiple hypoechoic areas. Contrast-enhanced abdominal computed tomography (Fig. 1) showed multiple hypodense areas, particularly in the upper pole of the spleen, covering 40% of the total volume, which is compatible with splenic infarcts.
 Axial section, portal phase. A hypodense, wedge-shaped hypocaptive image is seen, with a tip oriented towards the hilum in the anterosuperior aspect of the splenic parenchyma. B) and C) Multiple hypodense hypocaptive images: the largest wedge-shaped with a tip oriented towards the hilum in the anterosuperior aspect of the splenic parenchyma and the others smaller. D) Coronal reconstruction. Multiple hypocaptive images are evident, the largest of them located predominantly in the upper pole of the spleen, wedge-shaped in the splenic parenchyma and pointed towards the hilum, which covers 40% of the volume of the spleen corresponding to splenic infarcts.")
Contrast-enhanced abdominal tomography. A) Axial section, portal phase. A hypodense, wedge-shaped hypocaptive image is seen, with a tip oriented towards the hilum in the anterosuperior aspect of the splenic parenchyma. B) and C) Multiple hypodense hypocaptive images: the largest wedge-shaped with a tip oriented towards the hilum in the anterosuperior aspect of the splenic parenchyma and the others smaller. D) Coronal reconstruction. Multiple hypocaptive images are evident, the largest of them located predominantly in the upper pole of the spleen, wedge-shaped in the splenic parenchyma and pointed towards the hilum, which covers 40% of the volume of the spleen corresponding to splenic infarcts.
Cardiac and septic embolism were ruled out, as well as hematological disorders, malignancies, and deep vein thrombosis. No signs of pulmonary thrombosis were found in angiotomography, and the presence of valvular disease or vegetations was excluded in the transthoracic echocardiogram.
Immunological studies, aimed at evaluating the presence of APS as an etiology of splenic infarcts, showed high levels of anticardiolipin IgG (128 GLP U/ml) and beta-2 glycoprotein IgG antibodies (>100 U/ml), while the lupus anticoagulant was negative.
Parenteral pulsed methylprednisolone was started for 3 consecutive days, and once the diagnosis of APS was confirmed, anticoagulation with heparin followed by coumarins was started. The patient had a favorable evolution; she was discharged, and prednisone, hydroxychloroquine, and warfarin were prescribed.
DiscussionAPS is an autoimmune disorder characterized by a hypercoagulable state that can result in recurrent venous thrombosis or arterial occlusive events and fetal losses associated with elevated levels of antiphospholipid antibodies5,6; likewise, it can constitute a primary condition or occur in the context of autoimmune diseases, especially SLE,1,2,5 as occurred with our patient.
Antiphospholipid antibodies promote the inhibition of the anticoagulant cascade, which promotes thrombosis. Endothelial cells, monocytes, platelets, and complement activation pathways play a central role in SAF-induced arteriovenous thrombosis and embolism.6
Most clinical signs of APS are related to recurrent venous, arterial, or small vessel thromboses that occur in any tissue or organ.7,8 These thromboses can be single episodes, without complications, or they can constitute potentially fatal situations of massive, recurrent, and generalized thromboembolic events, which is known as catastrophic APS.9
Among the clinical manifestations of APS in adults, deep vein thrombosis remains the most frequent (39%, -and may be accompanied by pulmonary embolism-), while ischemic cerebrovascular disorders and thrombocytopenia represent around 30% of cases. APS can also be associated with fetal loss in around 50% of pregnancies.9 However, thromboses can also develop in any other vascular territory, the most reported those that affect the liver,10 the kidney, the adrenal gland, or the heart.5 Splenic infarcts, evidenced in the case under discussion, are even rarer and are frequently described as part of catastrophic APS.11,12
Although APS can indeed be associated with autoimmune diseases such as SLE, splenic infarction as the first thrombotic manifestation has rarely been reported.13,14
Another unusual aspect within the presentation of APS is the association of hemolytic anemia with the thrombotic event. Vianna et al.15 identified in a series of cases that subjects with APS secondary to SLE and primary APS had similar clinical spectrums, although hemolytic anemia, low C4 levels, and neutropenia appear to be more frequent in individuals with APS associated to SLE.
Hemolytic anemia is related to an increase in thrombotic events.16 It has been postulated, based on animal models, that hemolysis can trigger the activation of procoagulant factors and alter erythrocyte morphology as a possible mechanism of thrombotic events.17
The diagnosis of APS is based on clinical and laboratory findings (Table 2) and is defined by at least one of two clinical criteria and one of two laboratory results (anticardiolipin IgM or IgG antibodies, lupus anticoagulant, or anti-β2 glycoprotein 1 antibodies).7,12 In the current case, the diagnosis was based on the presence of tomographically confirmed splenic infarction, a history of premature birth due to preeclampsia, and two laboratory criteria: anticardiolipin and anti-β2 glycoprotein 1 antibodies.
Diagnostic criteria for antiphospholipid syndrome.
| Clinical criteria |
|---|
| Vascular thrombosis |
| One or more episodes of arterial, venous, or small vessel thrombosis in any organ or tissue of the body, confirmed by appropriate imaging tests and/or histopathological analysis (thrombosis must be present without evidence of small vessel inflammation) |
| Morbidity during pregnancy |
|
| or |
|
| or |
|
| Laboratory criteria |
|---|
| 1. Lupus anticoagulant (LA) determined according to the recommendations of the International Society of Thrombosis and Hemostasis |
| 2. IgG and/or IgM anticardiolipin (aCL) antibodies measured by ELISA at medium or high titers (>40 GPL or MPL or >99th percentile) |
| 3. Anti-β2 glycoprotein 1 (aβ2GP1) measured by ELISA at medium or high titers (>99th percentile) |
| A definitive diagnosis of APS will be considered when at least one clinical criterion plus one laboratory criterion is present for two occasions, separated by an interval of at least 12 weeks. |
Source: Adapted from Ruiz-Irastorza et al.7
Splenic infarction may be an underestimated cause of acute abdominal pain. Its prevalence is close to 0.016%,1 and, in some case series, gastrointestinal symptoms are the first manifestations of APS.18 Splenic infarcts are reported between 1.1% and 1.8% of patients with APS.5,19
In Latin America, reports of splenic infarctions have been associated with the presence of sickle cell anemia in individuals of African descent, usually exposed to altitude.20–23 In the current report, no recent travel history, nor personal or family history of hemoglobinopathies, was identified. Splenic infarcts related to other hematological disorders24 and infections25 have been reported less frequently.
Ultrasound studies of the spleen continue to be very useful in the initial approach, although abdominal computed axial tomography usually provides the diagnosis since it allows the observation of hypodense images characteristic of a splenic infarction.2,3,8 Kaushik et al.26 showed that 2.8% of subjects with APS had splenic infarcts on abdominal tomography. With this technique, the “wedge” shaped lesions directed toward the capsule, as well as the apex directed towards the hilum, are suggestive of infarction.20 In the context of extensive necrosis due to liquefaction, not only a wedge-shaped hypodense, hypocaptive lesion can occur, but there can also be pneumatosis.26 In the current case, the lesions are in the upper pole and involve 40% of the volume of the spleen.
The treatment of choice for splenic infarction depends on the etiologies and is normally conservative. Therapeutic options are still a topic of discussion, although prolonged oral anticoagulation is generally accepted.7,8,27,28 Splenic infarctions do not usually require any surgical intervention, and splenectomy is only justified when complications appear such as splenic abscesses, ruptured spleen, hemoperitoneum, or persistence of pain.26
Individuals with splenic infarction may develop functional asplenia, depending on its extent, which would be suggested by the presence of Howell Jolly bodies and vacuolated red blood cells in the peripheral blood smear, and would be confirmed by scintigraphic studies of labeled erythrocytes with radioisotopes (99mTc) with Single Photon Emission Computed Tomography (SPECT).29,30 Given this confirmation, it should be considered to complement the vaccination scheme and protect against capsuled microorganisms.29
In conclusion, the case demonstrates the need to include splenic infarction associated with APS in the differential diagnosis of acute abdominal pain in patients with autoimmune diseases. Although rare, a splenic infarction may be an indication of the presence of APS, especially in patients with catastrophic APS.
Ethical aspectsThis case report has the patient's informed consent and her data were treated confidentially in the description of the manuscript. In turn, it has approval from the institutional ethics committee of the Hipólito Unánue National Hospital, Lima, Peru. Letter n°.045-2022-CIEI-HNHU.
FinancingNone.
Author contributionsJA and AS participated in the creation of the idea. JA, LC, and AS participated in the patient's clinical care and the collection of medical chart information. All authors participated in the preparation of the manuscript and its critical review and approved its final version.
Conflict of interestsThe authors declare that they have no conflict of interest in the preparation of this manuscript.
