




Interstitial Lung Disease Associated with Autoimmune Diseases
Más datosInterstitial lung disease (ILD) is a leading cause of both morbidity and mortality in systemic sclerosis (SSc). Radiographic lung abnormalities on high-resolution computed tomography (HRCT) imaging may be identified in 75–90% of those with SSc, while clinically significant ILD occurs in up to 40%. Early detection is important as early treatment in those with progressive ILD may improve outcomes. Appropriately risk-stratifying systemic sclerosis-associated ILD (SSc-ILD) is important in identifying those at highest risk of progression. This article summarises recent advances in SSc-ILD, particularly recommendations for screening, defining disease progression and monitoring.
La enfermedad pulmonar intersticial (EPI) es una de las principales causas de morbimortalidad en la esclerosis sistémica (SSc). Las anomalías pulmonares radiográficas en la tomografía computarizada de alta resolución pueden identificarse en el 75-90% de las personas con SSc, mientras que la EPI clínicamente significativa ocurre hasta en el 40%. La detección temprana es importante, ya que el tratamiento oportuno de los pacientes con EPI progresiva puede mejorar los resultados. Es importante estratificar adecuadamente el riesgo de EPI asociada a la esclerosis sistémica (SSc-EPI) para identificar a las personas con mayor riesgo de progresión. Este artículo resume los avances recientes en SSc-EPI, en particular las recomendaciones para la detección, la definición de la progresión de la enfermedad y el seguimiento.
Systemic sclerosis (SSc) is a heterogenous autoimmune disease involving multiple organ systems, characterised by the triad of fibrosis, inflammation and vasculopathy.1 SSc confers an increased risk of mortality, with a standardised mortality ratio of 2.7–3.4 compared to the general population,2,3 and more than 20 years of life lost.3 Cardiopulmonary complications, particularly interstitial lung disease (ILD) and pulmonary arterial hypertension (PAH), are a major contributor to mortality in SSc.3 SSc-ILD is implicated in 20–35% of SSc-related deaths,2–4 and tends to occur early in the disease course, particularly in the first 5 years from symptom onset.5,6
ILD in SSc is highly prevalent, although exact estimates depend on the diagnostic modality used. Up to 75–90% of individuals with SSc will display some abnormality on chest high-resolution computed tomography (HRCT),7,8 the gold-standard diagnostic investigation for SSc-ILD.5 While some individuals may have subclinical disease, around 40% will develop moderate or severe disease.9 Thus, there is a broad spectrum of disease in SSc-ILD, and a variable natural history. Identifying those at highest risk of progressive ILD is important in determining optimal management, as early initiation of treatment to prevent further loss of lung function is likely to improve survival. Here, we aim to summarise current knowledge about SSc-ILD, with emphasis on diagnostic approaches and risk-stratification.
Who is most at risk of ILD in SSc?Patients with scleroderma at increased risk for developing ILD include those with diffuse cutaneous SSc (dcSSc),6,10,11 male sex12 and anti-Scl-70 antibodies.6,11,12 Certain racial groups have also been identified as being at higher risk of developing SSc-ILD, particularly individuals of African-American13 and Asian14,15 ethnicity. People with gastro-oesophageal reflux disease (GORD) also appear to be at higher risk of SSc-ILD.16 Anti-Scl-70 positivity has been associated with a higher risk of both developing ILD, and more severe ILD on both chest HRCT17 and as measured by FVC values lower than 70%.10,17 Anti-centromere antibody (ACA) positivity confers a lower risk of developing ILD.12 Data are conflicting regarding ILD risk in those with anti-RNA polymerase-3 (RNAP3) positivity; while some data suggest around 50% of RNAP3-positive individuals may present with ILD,18,19 others suggest a lower frequency and less severe ILD in this group.20,21 In the two studies reporting 50% ILD prevalence in RNAP3 positive participants, this group tended to have milder disease at baseline.18,19
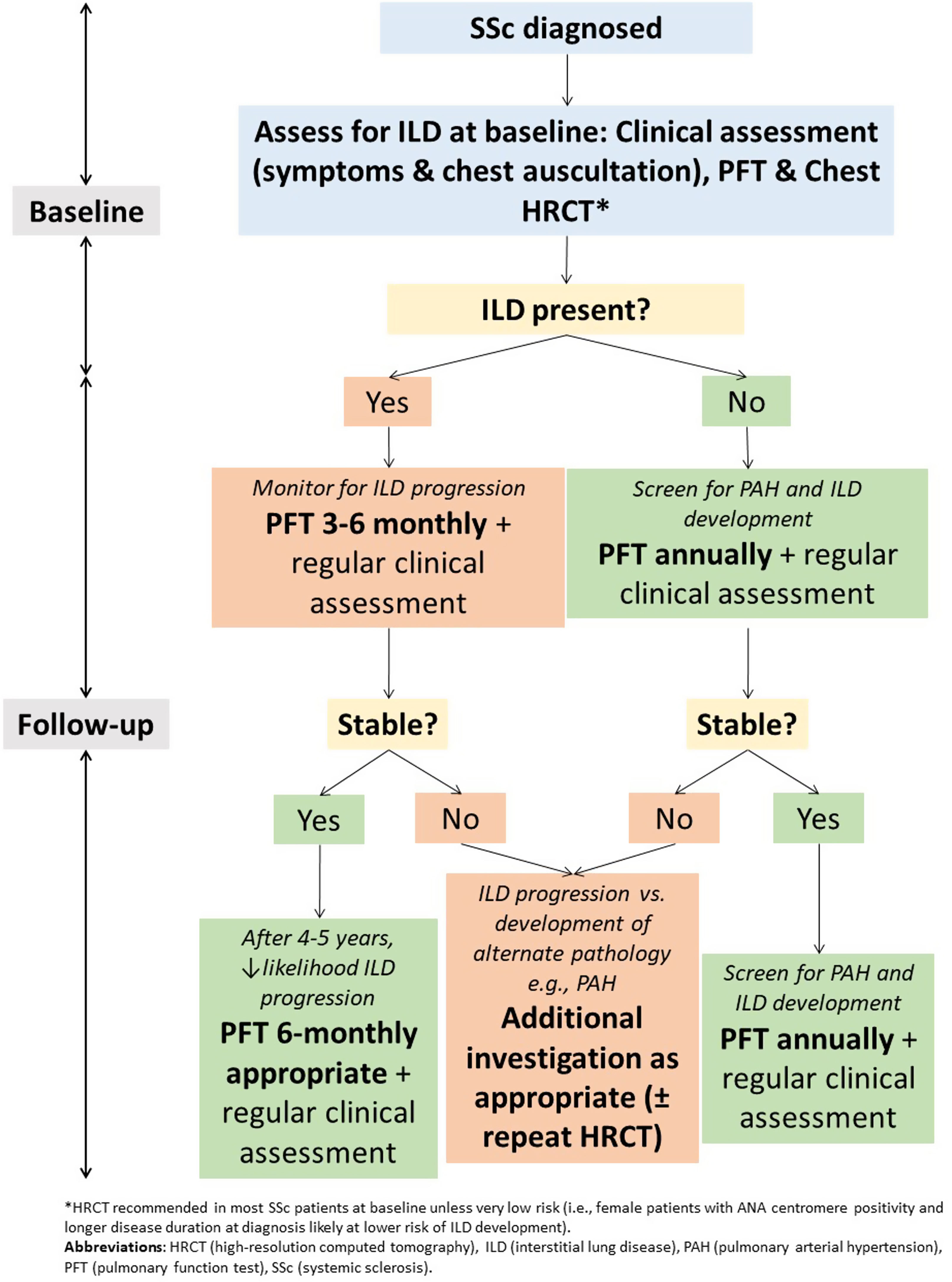
How should we screen for SSc-ILD?Screening for ILD is recommended for all patients with SSc and should include assessment of symptoms, chest auscultation, spirometry with DLCO, chest HRCT at baseline, and/or autoantibody testing.22 The presence of respiratory symptoms, such as exertional dyspnoea and cough, is important in the screening of SSc-ILD, although the use of symptoms alone is unreliable. Respiratory symptoms are often insidious in onset and can be masked by other factors such as relative immobility and pain from musculoskeletal disease. Pulmonary function testing (PFT) remains an important component of screening for, and assessment of, both PAH and ILD in SSc. While FVC is predominantly a marker of the pulmonary interstitium, DLCO is a marker of both the pulmonary interstitium and vasculature. The utility of PFTs is limited by the wide range of normal values. Indeed, up to 60% of individuals with ILD evident on chest HRCT may have normal forced vital capacity (FVC).23 Chest HRCT remains the gold standard for the detection of ILD and is more sensitive than chest X-ray imaging.5,23,24 The radiographic extent of fibrosis at baseline may be associated with FVC decline, with greater than 25% extent of fibrosis on baseline HRCT associated with increased rate of FVC decline.25
HRCT staging systems also provide important prognostic information. Radiographic extent of ILD on baseline HRCT is associated with increased mortality, especially with greater than 20% total lung involvement, or 10–20% involvement together with an FVC lower than 70%.26,27 Moreover, a normal chest HRCT at baseline confers a lower risk of developing SSc-ILD in future, and thus is associated with a better prognosis.28,29 In one study, 85% of people with a normal chest HRCT at baseline did not develop ILD over 5 years.28 Given that SSc-ILD most commonly develops in the first 5 years of SSc onset, performing chest HRCT around the time of diagnosis seems to be an optimal timepoint.5,6
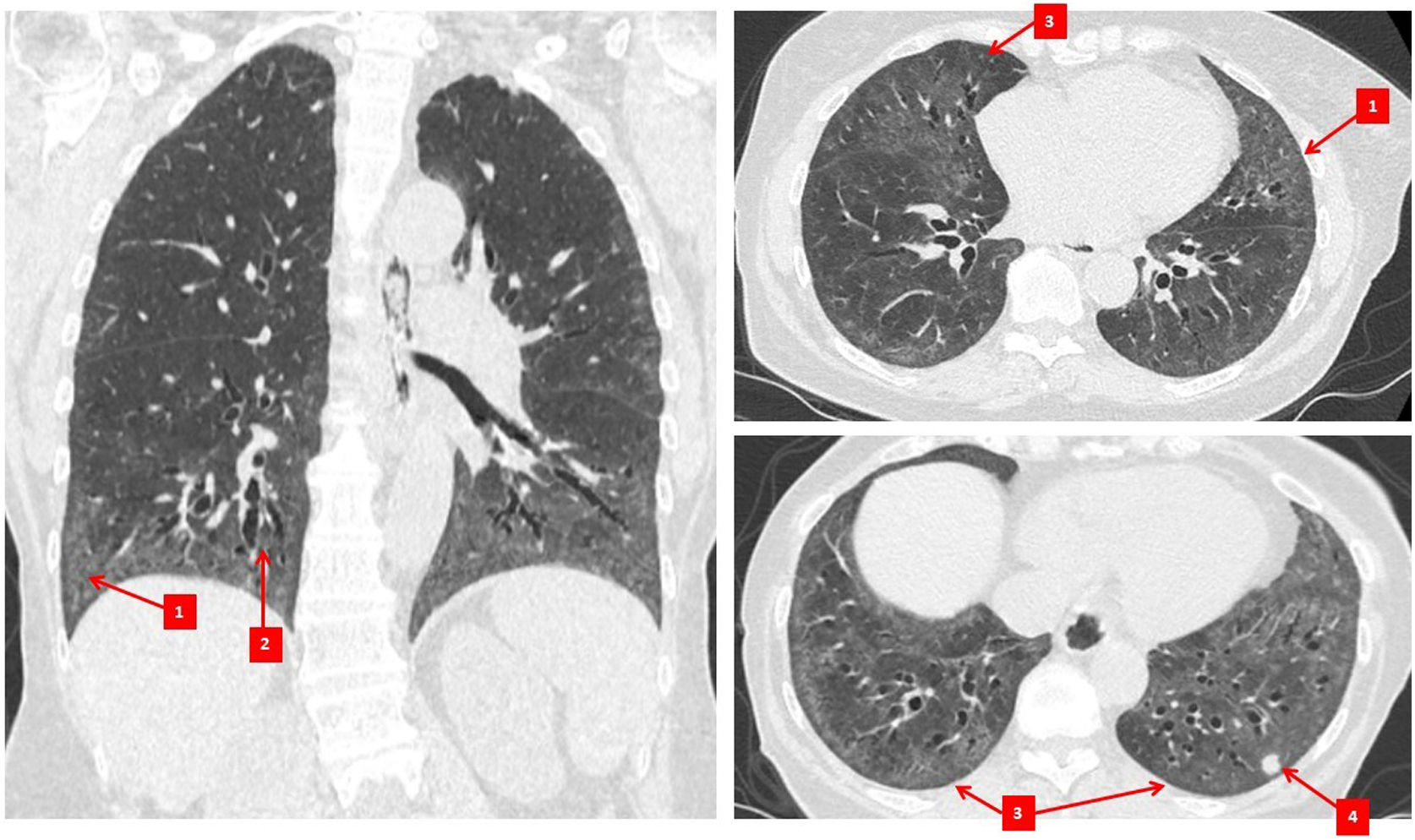
Diagnosis and monitoring of SSc-ILDImaging studiesChest HRCT imaging is now considered the gold standard investigation for ILD diagnosis in SSc.5,23 The most common radiographic pattern of involvement is non-specific interstitial pneumonia (NSIP), occurring in 70–80% of patients5,30,31 (Fig. 1), while between 10 and 25% of patients will have a usual interstitial pneumonia (UIP) pattern.30,31 NSIP is characterised by the presence of a mixture of reticular and ground glass changes, whereas UIP is characterised by the presence of honeycombing (the cardinal feature) and reticular changes with minimal ground glass opacities.32 While ground-glass opacities may be an indicator of active inflammatory disease/alveolitis, they can also be a reflection of fine fibrosis, especially if the changes are adjacent to areas of traction bronchiectasis.33 This is exemplified by the discordance in the frequency of ground glass opacities on CT34 compared to the frequency of active alveolitis in histopathologic studies in SSc-ILD.30
 Ground glass opacification, (2) traction bronchiectasis, (3) subpleural sparing, and (4) lung nodule.")
Another benefit of chest HRCT in SSc is the ability to identify other pulmonary pathologies. For example, SSc and SSc-ILD may be associated with an increased risk of lung cancer,35 which may be identified on chest HRCT imaging, although best assessed on contrast-enhanced standard chest CT imaging. Chest HRCT imaging may identify an enlarged pulmonary artery suggestive of PAH.36,37 Furthermore, bronchiectasis may occur more frequently in SSc and is readily detected by chest HRCT.38 Bronchiectasis appears to occur independently of ILD,38 suggesting this is a separate pathologic entity which can contribute to dyspnoea, cough and recurrent infections. Additionally, a proportion of individuals may have combined pulmonary fibrosis and emphysema identified on imaging.39,40 Emphysematous change can occur in SSc in non-smokers; 62.5% of participants had never smoked in one study of people with SSc with emphysematous change on surgical lung biopsy.41 Combined emphysema and fibrosis is associated with a lower diffusing capacity for carbon monoxide (DLCO),40,41 higher radiographic extent of fibrosis39,41 and in one study, small vessel vasculopathy in the pulmonary arteries.41 It is important to identify this emphysematous change as the consequential reduction in DLCO may confound screening for PAH, and may also contribute to more severe respiratory disease including a higher risk of pulmonary hypertension.39
The role of chest HRCT in monitoring progression of SSc-ILD is not yet fully defined. While experts agree that HRCT can be useful in monitoring ILD progression,42 there is no agreement on how frequently it should be performed. Recent consensus guidelines suggest chest HRCT should be performed according to clinical need, based on the current symptoms, disease state, and the speed of progression.42 Observational data suggest that most patients with a baseline extent of fibrosis on chest HRCT of <20% continue to remain stable, with a minority (especially those with shorter disease duration) progressing to >20% CT extent.29 Other data suggest that greater than 25% baseline HRCT extent of fibrosis has been associated with increased FVC decline.25 Changing HRCT pattern or extent may also be helpful to monitor the impact (or lack thereof) of therapy.43 Furthermore, changes in HRCT pattern may correlate with other symptom and functional outcomes, with reduced dyspnoea in those with improving quantitative interstitial lung disease scores identified in a subgroup analysis of the Scleroderma Lung Study II.44 Further data are required to understand the optimal use and timing of serial chest HRCT imaging in SSc-ILD.
Currently, exposure to ionising radiation is a major concern limiting routine repeat use of chest HRCT.45 Younger age confers increased risk from exposure, with the highest risk in children and young adults.46 The estimated radiation exposure from chest HRCT scanning is 2–4mSv,47 compared to standard CT chest imaging of 7mSv48 or low-dose lung cancer CT screening of 1.4mSv.49 For broader context, annual background radiation exposure worldwide is estimated to be 2.4mSv per annum,50 and the World Health Organisation suggests a threshold lifetime exposure potentially associated with increased cancer risk of 100mSv.46 Recent technological advances may allow lower-dose CT protocols to be used in ILD monitoring.45,51 Low-dose and ultra-low dose CT imaging techniques have been used in other contexts, for example lung cancer screening52 and monitoring of pulmonary nodules,53 although these have been more difficult to apply in ILD. Advanced image reconstruction methods, including iterative reconstruction techniques, use of limited axial scans (using thin slices over large intervals e.g., every 10–20mm), using detector systems with increased efficacy and automatic exposure control are all strategies to reduce radiation exposure.45 Lower dose chest HRCT scanning is likely to be a valuable tool in SSc-ILD, particularly in monitoring after an initial diagnosis.
No other imaging investigation is currently routinely recommended in clinical practice for diagnosis, screening or monitoring of SSc-ILD. However, much interest in other modalities has arisen in an attempt to minimise radiation exposure, particularly in monitoring SSc-ILD where serial testing may be required. Ultrasound imaging of the lung may be able to identify features suspicious for ILD.45 In particular, ultrasound detection of multiple “B-lines”, pleural irregularity and pleural thickening can predict HRCT diagnosis of SSc-ILD.54,55 Magnetic resonance imaging (MRI) may give some information about inflammatory changes in the lung in pulmonary fibrosis.56,57 However, chest HRCT currently provides superior imaging quality compared to lung MRI.45 Positron emission tomography combined with CT (PET-CT) may identify increased 18FFDG (Fluorodeoxyglucose F18) uptake within the lungs in myositis-associated ILD, which may help both in diagnosis and to identify those at risk of rapidly progressive ILD.58 Further data are required in SSc cohorts to identify the role of these imaging modalities in SSc-ILD in both diagnosis and monitoring, especially where ionising radiation exposure may be lessened or avoided.
Pulmonary function testingWhile limitations exist when PFT are used in isolation as a screening or diagnostic test for ILD, they remain important in monitoring of SSc-ILD (Fig. 2). This is particularly true given the exposure to ionising radiation with serial HRCT examinations. PFT convey important prognostic information and are important in assessing ILD severity and defining clinically significant disease.42 It is recommended that PFT are repeated every 3–6 months in those with ILD, with more frequent testing earlier in the disease course where risk of progression is highest59 (Fig. 2). In those without ILD, PFT can be monitored annually to screen for both ILD and PAH.

Small declines in FVC and DLCO are associated with reduced survival.59–61 The estimated clinically important difference for percent-predicted FVC is only 2–6%, with an estimated test–retest variability of PFT measurements of around 5–10%.59,62 Risk of progression is highest in the first 4–5 years of ILD, where close monitoring is recommended.59 In one study, those who maintain stable PFT for around 4 years post-ILD diagnosis were at lowest risk of ILD progression.59 However, stability of FVC over one time interval does not guarantee future stability. Up to 30% of individuals with a previously stable FVC may experience significant deterioration in the subsequent 12 months,63 highlighting the need for serial PFT monitoring.
PFT in SSc provide important additional benefits, particularly in screening for pulmonary hypertension. The development of pulmonary hypertension, including precapillary PAH, has major prognostic and functional implications in SSc and can develop in those with SSc-ILD.15,29,64 PFT are a useful tool in screening for SSc-PH, particularly if DLCO declines out of proportion to change in FVC. Early identification of PAH is critically important and improves survival and haemodynamics.65–67 PFT results may help to identify anaemia where haemoglobin testing is performed to assess oxygen transfer, as well as extra-thoracic restriction related to severe skin involvement. Furthermore, PFT can also assess for respiratory muscle weakness in those with SSc and myositis overlap syndromes.68 Accordingly, PFT continue to have an important role in monitoring SSc-ILD, assisting with prognostication, and in detecting other SSc complications or contributors to breathlessness.
Six-minute walk testingSerial six-minute walk testing (6MWT) may also be a helpful tool in monitoring individuals with SSc-ILD and PAH, with the combined utility of measuring both distance walked and detecting exertional desaturation using oxygen saturation on room air and with supplemental oxygen where appropriate.69 6MWTs can help to monitor functional exercise capacity in a standardised way, although the results can be affected by non-cardiopulmonary contributors to exercise intolerance (e.g., joint or muscle pathology) in SSc.69 Those with SSc-PAH may have shorter six-minute walk distances than those with SSc-ILD alone.69 However, the utility of 6MWT in monitoring SSc-ILD is unknown. In a meta-analysis of randomised controlled trial data in non-SSc cohorts with PAH, change in 6MWD measurements did not correlate with clinical outcomes (death, hospitalisation, or rescue PAH therapy).70
Invasive investigationsPreviously, surgical lung biopsy was commonly performed to investigate SSc-ILD and define the pattern of involvement.30 However, this has been largely superseded by HRCT imaging as a reliable and non-invasive tool to determine ILD subtype, unless exclusion of malignancy or an alternate pathology is required.71 Bronchoscopy and bronchoalveolar lavage (BAL) can be used both to exclude infection, and to guide prognosis and treatment in ILD. Higher neutrophil count on bronchoalveolar lavage has been shown to correlate with more extensive disease on CT, and more fibrotic disease.72 Inflammatory BAL results suggestive of alveolitis are associated with more severe restrictive ventilatory defects.73,74 However, the invasive nature of bronchoscopy and variable analytical techniques in processing of BAL samples has limited its widespread use.74 Furthermore, whether BAL results confer additional prognostic or therapeutic information beyond non-invasive testing is uncertain.74 It is possible that with the advent of novel biomarkers for SSc-ILD, analysis of specific biomarkers on BAL sampling may be beneficial in risk-stratification or determining the likelihood of treatment success.71,74
Multidisciplinary team discussion in SSc-ILD diagnosis and managementMultidisciplinary team (MDT) discussions play an important role in diagnosis75,76 and management of ILD.77 Whilst the diagnosis of ILD in the presence of confirmed SSc is usually clear, identifying disease progression and deciding when to adjust treatment can be more difficult. Furthermore, the clinical features of SSc can be subtle in a patient with de novo ILD. Consideration should be given to discussing those with SSc-ILD at a multidisciplinary meeting which includes the treating rheumatologist, along with a radiologist and respiratory physician. Studies suggest including the rheumatologist in this multidisciplinary meeting is beneficial; in one review, only 24% of general ILD multidisciplinary meetings included a rheumatologist.77 However, ILD multidisciplinary meetings have an important role in adjusting immunosuppression and increasing use of non-corticosteroid immunosuppression in ILD, particularly connective tissue disease (CTD)-associated ILD.78 Inclusion of a rheumatologist in multidisciplinary discussions has been shown to assist with modifying treatment in up to 80% of CTD-ILD cases.77,79
Defining ILD progressionNot all individuals with chest HRCT abnormalities develop progressive ILD, thus it is important that all patients with SSc-ILD are closely monitored, particularly early in the disease course. Monitoring for ILD progression should include regular assessment of symptoms and exercise tolerance, physical examination, and pulmonary function tests.80 While there are no current recommendations about the indications for, and timing of, serial chest HRCT examination, this may be considered in the case of progressive symptoms, to assess treatment response, and to investigate acute exacerbations.
There is no current universally accepted definition for SSc-ILD progression.81–83 However, the OMERACT CTD-ILD working group have proposed the following definition for clinically-meaningful progression in CTD-ILD, now widely accepted in SSc80,83; ≥10% relative decline in FVC (%) or ≥5 to –<10% relative decline in FVC (%) and ≥15% relative decline in DLCO (%).84 This definition is generally applied over 1–2 years.80 Radiographic progression is also an important indicator, where these data are available, although the minimum clinically important change in HRCT extent is yet to be defined. Some data suggest that early radiographic progression in SSc-ILD may be associated with worse survival. In a subgroup analysis of Scleroderma Lung Study I and II participants, ≥2% progression on chest HRCT using quantitative imaging analysis was associated with worse survival.85 Further data are required to optimally define SSc-ILD progression, and in particular radiographic progression on HRCT.
Can we predict who will develop progressive SSc-ILD?Predicting who will develop progressive ILD is critically important in optimising both the intensity of monitoring and treatment. While much work has been done to identify features associated with higher risk of ILD progression, this remains an imprecise science. Older age at SSc onset,12 dcSSc63,86 and male sex12 have both been associated with increased risk of ILD progression. People with ACA positivity are likely to be at lower risk of ILD progression28,29; ACA positivity has been associated with milder ILD60 and preservation of lung function.29,87 Although anti-Scl-70 positivity is associated with the development of ILD,6,11,12 its utility as a marker of progression is unclear. While some data suggest anti-Scl-70 positivity may be associated with ILD progression,12,88 other data have failed to identify an association between anti-Scl-70 positivity and either radiographic progression or an FVC decline of greater than 10%.17 Data are conflicting regarding RNAP3 positivity and ILD progression. In one study assessing radiographic progression of RNAP3 positive participants on HRCT, only 18% developed severe fibrosis after 3 years.19 In contrast, a greater proportion of participants with RNAP3 positivity progressed compared to those with anti-Scl-70 positivity.19 Gastroesophageal reflux disease (GORD) has also been identified as an important contributor to SSc-ILD progression.89,90 In one study the burden of reflux symptoms was associated with greater progression of SSc-ILD.89 This underscores the importance of aggressive anti-reflux treatment in those with SSc-ILD.
Markers of increased ILD severity at baseline have been shown to confer increased risk of progression. These include higher baseline radiographic extent of fibrosis,12,27,29 particularly greater than 20% extent of fibrosis on HRCT chest,27,29 reduced FVC levels,6,12 reduced DLCO levels6,12 and decreased oxygen saturation.87,91 Outcomes appear to be more closely linked to ILD severity at presentation and decline in PFT parameters rather than baseline HRCT pattern.30
Other tools in SSc-ILD: the promise of biomarkers in ILD diagnosis and risk stratificationWhile not yet routinely used in clinical practice, novel biomarkers detected in both serum and bronchoalveolar lavage fluid have been shown to be associated with both the presence and progression of SSc-ILD.92 CC chemokine ligane-18 (CCL-18) is a chemokine expressed by alveolar macrophages, while carbohydrate antigen 15.3 (CA15-3) and Krebs von Lungen-6 (KL-6) are both glycoproteins encoded by the gene MUC1.93 Interleukin-6 (IL-6) is a proinflammatory cytokine.94 Ca15-3,95 KL-6,92 IL-8,92 IL-696 and surfactant protein D92 may all be associated with the presence of ILD, while CCL-1812,97,98 and Ca15-399 may be associated with increased ILD severity. CA15-3 may also correlate with dyspnoea.99 CCL-1897,98 and CCL-2100,101 levels may be associated with survival, while KL-6 has also been associated with survival in cohorts with idiopathic pulmonary fibrosis.102 CCL-2,101 Ca15-3103 and KL-617,104 are associated with greater radiographic extent of ILD, while CCL-18,17,97,98,105 CCL-2,100,101 Ca15-3,99,106 KL-612,107,108 and surfactant protein D17,105,109,110 may correlate with PFT measurements. CCL-2,100,101 Ca15-399,106 and KL-6107,111 may be associated with ILD progression or PFT decline; data are conflicting about the role of IL-6 in progression.100,112 In one study, the use of a composite score including surfactant protein D, Ca15-3 and Intercellular Adhesion Molecule 1 (ICAM-1) was found to be more predictive of disease than individual markers alone.113 The utility of this composite score as a prognostic marker is currently unknown. Multiple other biomarkers are currently under investigation, and all need to be validated in large, multicentre, prospective studies involving different racial groups.114
Other contributors to respiratory symptoms in SSc-ILDDyspnoea is an important symptom of SSc-ILD, and an increase in dyspnoea is generally associated with progressive disease. However, several co-morbidities in SSc may also to contribute to this symptomology. Co-existing pulmonary hypertension is not uncommon in SSc-ILD and should be considered if there is a disproportionate drop in DLCO levels in relation to FVC levels. Data from the Australian Scleroderma Cohort study (ASCS), a large multicentre prospective cohort study of SSc, demonstrated that of 479 people with ILD, 106 (22.1%) also fulfilled RHC criteria for PAH.15 People with ILD combined with PAH have significantly worse physical function and survival compared to those with ILD alone.15 Concurrent left heart disease is common in SSc and left ventricular diastolic dysfunction (LVDD) occurs in 20–30% of people with SSc.115–117 Finally, anaemia and iron deficiency, skeletal muscle disease and respiratory muscle weakness, and fatigue can all contribute to reduced exercise tolerance and dyspnoea in people with SSc. Moreover, cough in SSc may also be multifactorial; post hoc analyses of the SSc Lung Study II data demonstrated that frequent cough was associated with more GORD symptoms.118 Accordingly, it is important to optimise anti-reflux treatment in anyone with SSc-ILD to avoid ILD progression,89 but also to optimise health-related quality of life and reduce symptom burden. A holistic approach and consideration of differential diagnoses is important in assessment of worsening respiratory symptoms in people with SSc-ILD.
ConclusionsILD is an important contributor to morbidity and mortality in SSc. There are well characterised clinical and serological factors that confer both an increased risk of ILD development and progression of ILD. Screening for ILD in SSc is recommended in all patients at baseline, and HRCT chest imaging is pivotal in the diagnosis of SSc-ILD. However, the role of chest HRCT in routine monitoring of SSc-ILD is yet to be established. PFTs remain the mainstay of ILD monitoring and should be performed every 3–6 months; more frequently during the first 5 years of disease where patients are at the highest risk of disease progression. Advances in novel biomarker discovery and other imaging modalities are likely to improve risk stratification and clinical care in SSc-ILD.
Conflict of interestJF has received conference sponsorship from Pfizer and honoraria from Boehringer-Ingelheim. MN has received honoraria or consultancies from Janssen, AstraZeneca, GlaxoSmithKlein, Boehringer-Ingelheim and Bristol-Myers Squibb. NG has received honoraria or consultancies from Boehringer-Ingelheim and Astra Zeneca, and an equipment grant from Air Liquide.
JLF holds a NHMRC Postgraduate ScholarshipGNT2013842 and an Australian Government Research Training Program Scholarship. MN holds an NHMRC Investigator Grant (GNT1176538).
The following are the supplementary material to this article:
