


El cáncer medular de tiroides familiar, neoplasia del tejido tiroideo poco prevalente, tiene su origen en una mutación del protoncogén RET. A continuación describimos un caso de presentación atípica y mutación poco frecuente (V804L), en el que la punción aspiración aguja fina y las técnicas de imagen no resultaron concluyentes y fueron las pruebas bioquímicas y el análisis molecular del gen RET las que permitieron llegar al diagnóstico y establecer el pronóstico del caso índice y sus familiares.
Familial medullary thyroid cancer (FMTC) is a non-predominant thyroid neoplasia originating from a proto-oncogene RET mutation. The case presented is atypical in its form of presentation, a fairly uncommon mutation (V804L) and does not have conclusive fine-needle aspiration biopsy (FNAB) and image studies. Biochemical and RET molecular analysis has a high diagnostic and predictive value in the index and familial cases.
El cáncer medular de tiroides familiar (CMTF) es una neoplasia poco frecuente de la glándula tiroidea, cuyo origen es el resultado de una mutación en el protoncogén RET. Puede presentarse aisladamente o asociada a otras endocrinopatías en el MEN21, mostrando un patrón autosómico dominante con penetrancia casi completa y expresividad variable2.
A continuación se describe un caso familiar de CMT de presentación atípica y mutación infrecuente3, resaltando la importancia de las pruebas moleculares en el diagnóstico y pronóstico del caso índice y sus familiares.
Caso clínicoMujer de 72 años que acude a consulta de digestivo por molestias abdominales y pérdida de peso en los últimos meses. Antecedentes personales sin interés. Alta prevalencia oncológica en antecedentes familiares (pulmón, gástrico, colon y próstata).
Todos los estudios bioquímicos y radiológicos (ecografía abdominal, gastroscopia y colonoscopia) resultan ser normales excepto la determinación de CEA de 40,7ng/ml. Se solicitan nuevas determinaciones hormonales (gastrina, VIP, catecolaminas, metanefrinas, GH, ACTH, insulina, glucagón, péptido C…). Estando todas ellas dentro de rango excepto la calcitonina de 858pg/ml, se decide realizar una interconsulta a endocrino ante la sospecha de tumor neuroendocrino vs. CMT.
Dentro de las pruebas de imagen realizadas, destaca la ecografía cervical que informa de 2 nódulos, uno en cada lóbulo tiroideo, de 1×2cm y 2×1,5cm, objetivándose mediante TAC cérvicotorácico calcificaciones intranodulares sin evidencia de linfadenopatías cérvicomediastínicas o axilares.
La punción aspiración con aguja fina bajo control ecográfico evidencia varios grupos celulares positivos para calcitonina y negativos para tiroglobulina no determinándose datos de atipia o malignidad celular, sin embargo, la insuficiencia y falta de representatividad de la muestra imposibilita el diagnóstico anatomopatológico de CMT.
La PET evidencia una captación compatible con bocio multinodular difuso y el rastreo gammagráfico con somatostatina (OCTREOSCAM) objetiva una asimetría tiroidea con aumento del lóbulo tiroideo izquierdo.
Una analítica posterior confirma elevación de calcitonina (1.134pg/ml) y CEA (76,2ng/ml).
Ante el diagnóstico de sospecha de CMT se solicita estudio de mutaciones en el protoncogén RET, detectándose una mutación germinal en heterocigosis que afecta al residuo 804 (V804L) en el exón 14.
Tras la confirmación del diagnóstico, se realiza tiroidectomía total con linfadenectomía cervical radical, evidenciándose en el estudio anatomopatológico posterior, proliferación neoplásica de células C y afectación ganglionar en 3 de los ganglios extirpados.
El seguimiento se realiza con calcitonina basal y tras estimulación con gluconato cálcico.
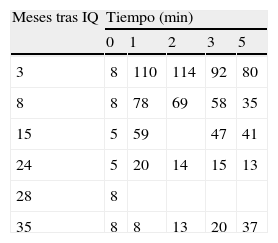
Ante la persistencia de niveles elevados de CT tras estimulación varios meses postintervención, se realiza gammagrafía tiroidea que confirma presencia de tejido tiroideo residual funcionante y se decide ablación con yodo radiactivo dado que el remanente es pequeño y de difícil extirpación. Como se observa en la tabla 1, los valores de calcitonina se normalizan (24 meses postintervención).
En la actualidad la paciente permanece estable.
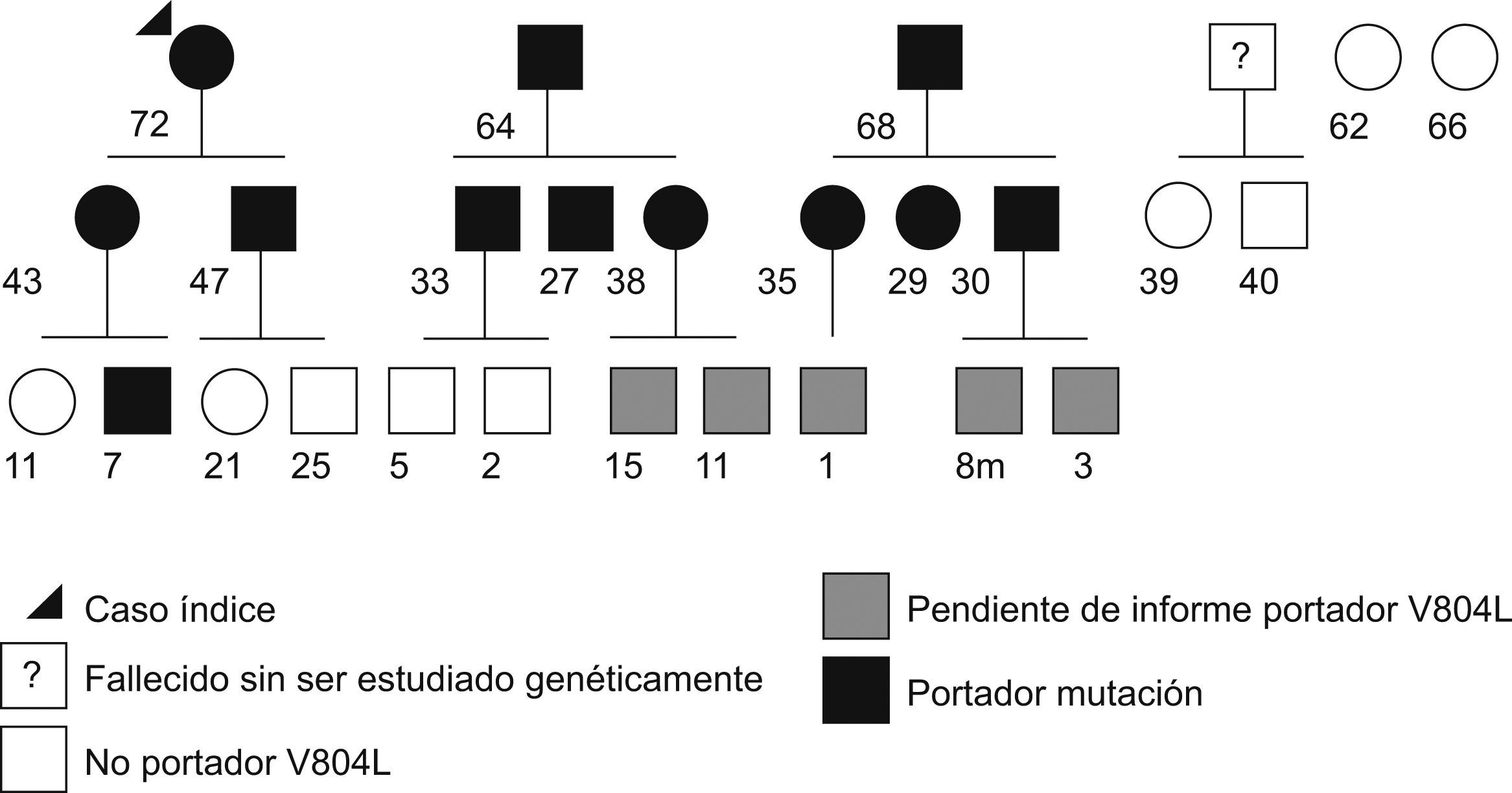
Debido a la alta penetrancia de la mutación y para determinar si se trata de un caso esporádico o familiar, se ofrece pruebas de cribado genético a familiares de primer grado por consanguinidad: 2 hermanos presentaron pruebas positivas, 2 hermanas negativas y en uno no se pudieron realizar por haber fallecido (fig. 1).

Los no portadores de la mutación fueron excluidos del protocolo de seguimiento. En los casos positivos se continuó el estudio molecular en la segunda generación, resultando portadores de la mutación los 2 hijos del caso índice y los 6 de sus 2 hermanos y no portadores, los 2 hijos del hermano fallecido.
Se continúa el estudio en la tercera generación, presentando pruebas positivas uno de ellos, negativas 5 y están pendientes de informe genético, otros 5 que no pertenecen a nuestra área sanitaria.
A todos los portadores de la mutación se les realizaron determinaciones de calcitonina basal y tras estimulación que resultaron ser anormalmente altas en el 33–55% de los casos respectivamente. Además para descartar el desarrollo de otras neoplasias asociadas a MEN2 se determinaron catecolaminas y metanefrinas en plasma y orina que resultaron normales en todos los casos.
Excepto un hermano del caso índice, todos los familiares que resultaron ser portadores optaron por la tiroidectomía como primera opción y en ellos se confirmó, mediante anatomía patológica, hiperplasia neoplásica de células C y CMT.
DiscusiónEl CMT constituye el 5–10% de las neoplasias de origen tiroideo1,4 y el único que puede ser diagnosticado por pruebas genéticas. Siendo la esporádica, la forma más frecuente de presentación (75%), resulta de especial interés la forma hereditaria, bilateral y multicéntrica5, cuyo origen es resultado de una mutación germinal en el protoncogén RET.
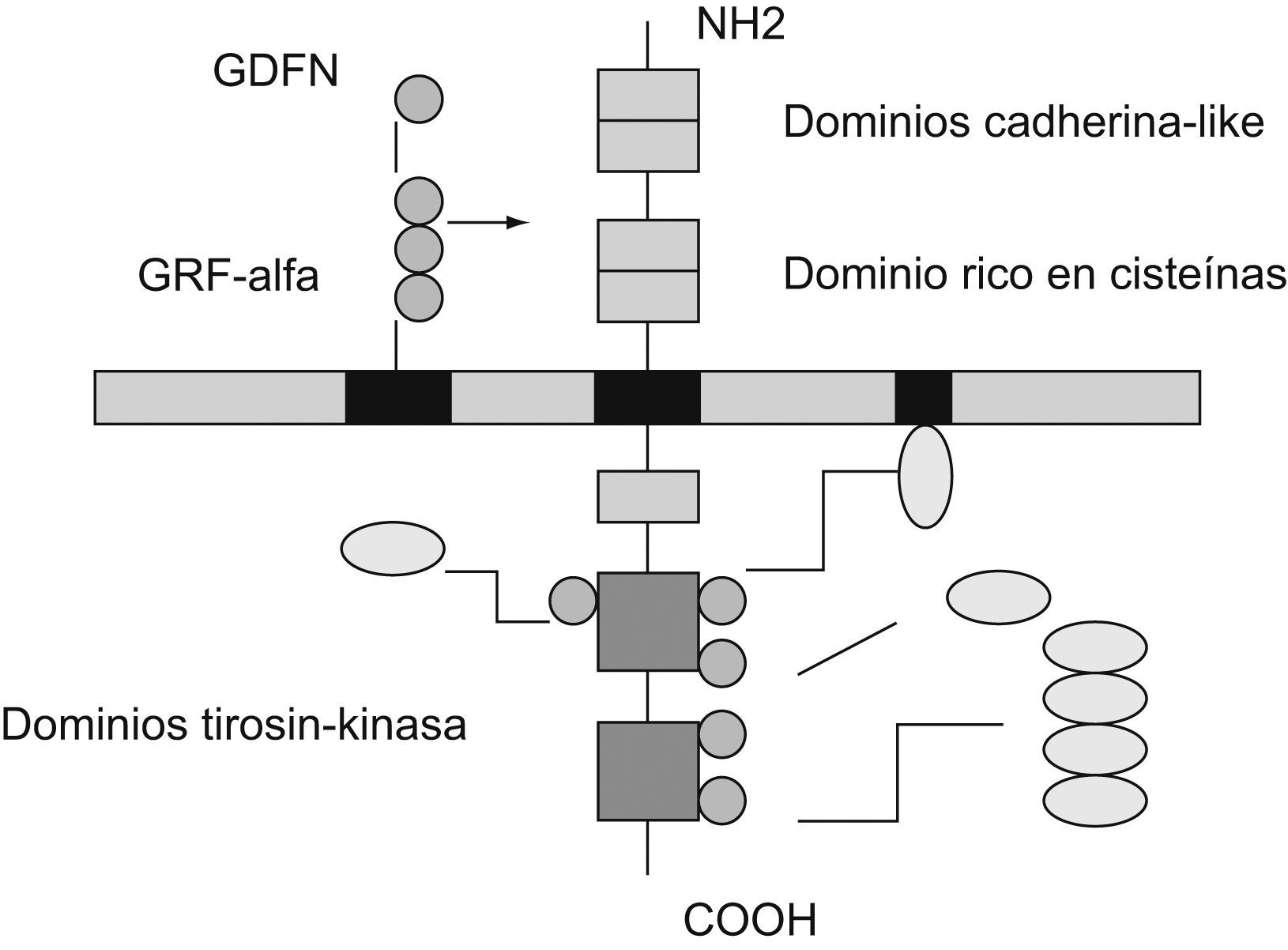
El protoncogén RET, situado en el cromosoma 10 (10q11,2), tiene 21 exones y codifica una proteína de la familia de los receptores de membrana con 3 dominios bien diferenciados (fig. 2): uno extracelular altamente conservado (péptido señal N-terminal, cadherin-like y dominio rico en cisteínas), otro transmembrana y otro intracelular (tirosin-quinasa)5,6 (fig. 2).

La proteína RET juega un papel esencial en la migración y desarrollo de los tejidos que surgen de la cresta neural. Está expresado en células C del tiroides, médula adrenal, paratiroides, ganglios simpáticos y parasimpáticos, ganglios entéricos y tracto urogenital6. Hay 3 isoformas del RET que desempeñan papeles distintos en la diferenciación tisular embrionaria7.
Son numerosas las mutaciones conocidas que producen sobreexpresión del protoncogén RET e incremento de la transmisión de señales dando lugar a proliferación celular. Dependiendo de la localización de la mutación se pueden clasificar7,8 en: mutaciones que afectan al dominio extracelular (exones 10 y 11), responsables del 98% de los casos MEN2A y CMTF y mutaciones que afectan al dominio intracelular, responsables del 4% de CMTF exones (13, 14 [V804L mutación de nuestra paciente] y 15) y asociadas exclusivamente a pacientes con MEN2B (exones 15 y 16).
El cribado de familiares de 1.er grado que clásicamente se realizaba mediante la determinación de calcitonina basal o tras estimulación9,10, está siendo sustituido por la detección genética de las mutaciones en el protoncogén RET ya que el estudio molecular se ha convertido en el gold estándar como prueba diagnóstica y predictiva del CMT, con sensibilidad y especificidad próximas al 100%11.
El caso descrito es un CMT de tipo hereditario, bilateral y multicéntrico, de presentación clínica atípica, sin disfunción tiroidea, crisis hipertensivas u otros síntomas neuroendocrinos (motivo de consulta: dolor abdominal y pérdida de peso) y una mutación poco frecuente (V804L).
Aunque CT y CEA resultan anormalmente elevados, las técnicas de imagen: ecografías, TAC, PET y octreoscam, así como la punción aspiración con aguja fina no son concluyentes por lo que el estudio genético del protoncogén RET fue determinante para el diagnóstico de CMT, confirmado por el estudio histológico posquirúrgico.
El CMT es la primera manifestación neoplásica en la mayoría de los individuos MEN2, de modo que algunas familias han sido incluidas incorrectamente como CMTF con el consiguiente riesgo de no prevenir el feocromocitoma12. Concretamente la mutación V804L ha sido descrita y asociada fundamentalmente con CMTF, pero algunas series han apuntado que puede relacionarse con el desarrollo de feocromocitoma. Las pruebas genéticas y bioquímicas fueron decisivas en el cribado familiar confirmándose que estamos ante un caso de CMTF con al menos 12 miembros portadores de la mutación, estando 5 pendientes de informe.
- •
Franquelo R, Jefe de Servicio Análisis Clinicos, Hospital V. de la Luz, Cuenca.
- •
del Río J.J, Servicio Anatomía Patológica, Hospital V. de la Luz, Cuenca.
- •
García A, Especialista en Inmunología, Hospital V. de la Luz, Cuenca.
- •
Dalmau, María.
