


La granulomatosis linfomatoide (GL) es un proceso angiocéntrico y angiodestructivo que frecuentemente afecta al pulmón en forma de infiltrados nodulares bilaterales compuestos de una población mixta de células linfoides que carecen de características granulomatosas verdaderas1.
Paciente de 65 años con antecedentes de inmunodeficiencia común variable en tratamiento con inmunoglobulinas parenterales cada 21 días, psoriasis e hipotiroidismo primario. Consulta por aumento del perímetro abdominal en los últimos días. Niega pérdida de peso, fiebre, ni otros síntomas B. No tos, hemoptisis, sangrado vaginal, ni cambios de ritmo intestinal. Niega episodios de ictericia o transfusiones. En pruebas complementarias presenta anemia (hemoglobina 10,2g/dl), mínimo aumento de GGT y FA, con el resto de hemograma, bioquímica, serologías y marcadores tumorales dentro de la normalidad, salvo beta-2-microglobulina en 9,3mg/l. La TAC mostraba derrame pleural (aspecto maligno), nódulos pulmonares sospechosos de metástasis, esplenomegalia con múltiples LOEs, adenopatías de aspecto patológico mesentéricas, retroperitoneales y axilares derechas; marcada ascitis. Pequeña tumoración en uréter proximal del riñón izquierdo y engrosamiento de pared uterina. No lesiones cerebrales. En la PET se confirma afectación adenopática supra e infradiafragmática (SUVmáximo 38,05), con infiltración hepatoesplénica de médula ósea y pulmonar.
El estudio ginecológico y urológico descartaron tumores primarios de dicho origen, por lo que se biopsió una adenopatía axilar. Previamente se realizó endoscopia digestiva alta, cuya biopsia ya fue sugestiva de proceso linfoproliferativo B.
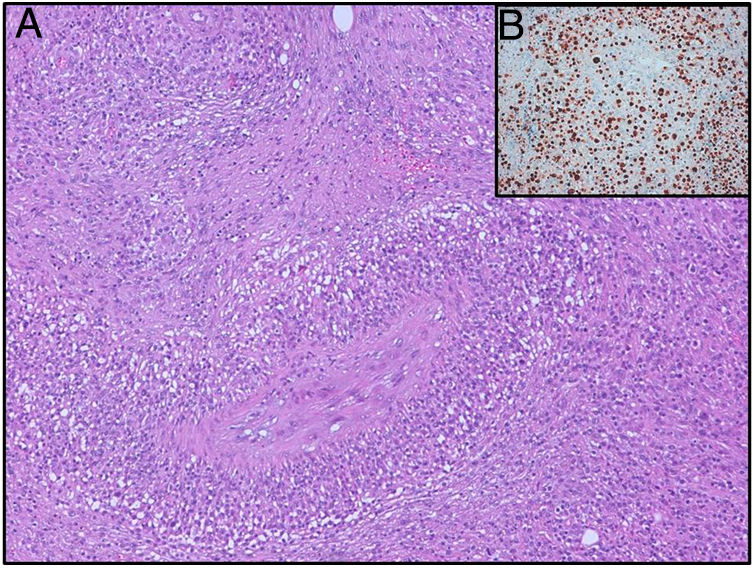
El ganglio linfático estaba sustituido por una población linfoide de características blásticas con citoplasma amplio y claro, con límite definido, núcleos con nucléolo prominente y abundantes formas sternbergoides que mostraban una disposición angiocéntrica, con áreas parcheadas de necrosis entremezcladas y una población de linfocitos pequeños acompañantes predominantes (fig. 1A). La celularidad tumoral se extendía al tejido adiposo perinodal formando nódulos no confluentes. Las células neoplásicas se teñían con intensidad frente a marcadores B (CD20, CD79a y PAX5) e inmunoexpresaban Bcl-2, CD30 y MUM-1, y eran negativas para Bcl-6, CD10 y CD15. Se acompañaba de una población de linfocitos T maduros CD3 positivos, con predominio de CD8, aunque existiendo también CD4 entremezclados. Se hicieron resaltar 2 aspectos importantes: primero la presencia de virus de Epstein-Barr (VEB) mediante inmunohistoquímica para el marcador LMP-1, como por CISH, en forma de latencia tipo II (EBER, fig. 1B) y, en segundo lugar, la marcada angioinvasión, producto de la cual existía la necrosis parcheada ya referida. Atendiendo a la clínica de la paciente, en lo que hace referencia a su inmunodeficiencia de base y la existencia de nódulos pulmonares múltiples en la TAC, se concluyó el previsible diagnóstico de GL.
 Población linfoide de características blásticas con nucléolo prominente y abundantes formas sternbergoides con disposición angiocéntrica entremezcladas con linfocitos pequeños acompañantes (HE, 4x). B) VEB positivo mediante CISH (EBER, 20x).")
Ante el diagnóstico de GL sistémica, se inicia tratamiento quimioterápico con esquema CHOP-rituximab (ciclofosfamida, adriamicina, vincristina y prednisona), obteniéndose escasa respuesta con un deterioro progresivo de la paciente, produciéndose el éxitus.
La GL fue descrita por primera vez por Liebow et al.2 en 1972, inicialmente se creía que se trataba de una enfermedad linfoproliferativa de células T, basada en el predominio de células T. Posteriormente Katzenstein et al. hicieron mención a su asociación al VEB. En 1994, Guinee et al. demostraron que el VEB estaba presente en las células B atípicas, en las cuales eran clonales y en algunos casos fue demostrada su clonalidad mediante reordenamiento para inmunoglobulinas2-5.
El diagnóstico diferencial hay que hacerlo con diferentes lesiones que muestran aspectos histológicos, inmunohistoquímicos similares, así como positividad para EBER entre las cuáles se encuentran trastornos linfoproliferativos iatrogénicos, postrasplante y asociados a inmunodepresión, infección? por HIV, linfoma de Hodgkin y linfoma de células T o NK4-6.
En la práctica el diagnóstico de GL es retante debido a que la enfermedad es extremadamente rara y las características histológicas pueden ser muy sutiles. Clínicamente, los pacientes deben tener afectación pulmonar, usualmente lesiones múltiples con predilección por lóbulos inferiores. En una cuarta parte de los casos se asocia a afectación del SNC, pudiendo dar lugar a una constelación de síntomas, si bien lo más frecuente es que se presente en forma de accidente cerebral vascular. La afectación extrapulmonar es común y está relacionada con progresión de la enfermedad. Existen pocos casos descritos con lesiones a nivel gastrointestinal7.
