


El cáncer renal tiene una incidencia del 3% del total de las neoplasias en adultos, y puede ser tanto de origen hereditario como esporádico. Histológicamente se clasifica en 5 tipos principales. Las técnicas actuales de citogenética y biología molecular han permitido conocer con más detalle algunos de los sucesos genéticos iniciales que causa la patogenia de los tipos mayoritarios como, por ejemplo, que un defecto en el cromosoma 3p está implicado en el desarrollo del carcinoma de células claras (ccRCC) o que en un 13% de los RCC de tipo papilar está mutado el gen c-MET (cromosoma 7), y en el resto de cánceres de este tipo existe duplicación de este mismo cromosoma, o que, en los oncocitomas la pérdida de los cromosomas 1 y/o 14 podría representar el suceso inicial. Aparte de estos defectos, el conocimiento de otros genes y alteraciones cromosómicas implicados, junto con otros factores, permite definir de manera aproximada el pronóstico en cada caso. Debido a que este tipo de cáncer desarrolla metástasis rápidamente, las terapias actuales, como la cirugía y el uso de interleucina (IL) 2, son poco eficaces y sólo consiguen alargar en unos meses la supervivencia del paciente. Aunque se están ensayando nuevas terapias, es necesario un conocimiento más profundo de los procesos moleculares implicados para obtener el éxito deseado tanto en el diagnóstico precoz como en el pronóstico y el tratamiento de la enfermedad.
Renal cancer has a 3% incidence of all neoplasms in adults and it can be of hereditary or sporadic origin. Histologically, it is classified into 5 main types. The current cytogenetic and molecular biology techniques allow some of the initial genetic events responsible for the main types of pathogenesis to be studied in more detail, such as the implication of a defect in chromosome 3p in the development of clear cell renal cell carcinoma (ccRCC), or the presence of the mutated c-MET gene (chromosome 7) in 13% of papillary RCCs and the presence of the same chromosome duplication in the rest of these types of cancer, or the loss of chromosomes 1 and/or 14 which could represent the initial event in oncocytomas. Apart from these defects, knowledge other genes involved and chromosomic defects, as well as other factors, may enable an approximate prognosis to be made in each case. As the metastatic process in this cancer type develops very early, the current therapies, such as surgery and the use of IL-2, are not very effective and they only manage to extend patient survival by a few months. Although newer therapies are being assessed, a deeper knowledge about the molecular processes involved is necessary to obtain the desirable success both in the early diagnosis, as well as the prognosis and treatment of the disease.
A pesar de los progresos alcanzados en el entendimiento de su biología, el carcinoma de células renales (RCC) se encuentra entre los primeros 10 tipos de cáncer que más muertes causan en la actualidad, y representa alrededor del 3% de los tumores sólidos que se presentan en adultos. Afecta 2 veces más a los varones que a las mujeres, y la incidencia es mayor entre los 50 y los 70 años. Se trata de una enfermedad muy heterogénea, y puede presentarse tanto de forma hereditaria como esporádica.
Gracias a los modernos métodos de imagen, como las ecografías, las tomografías computarizadas (TC) o la resonancia magnética (RM), el tumor puede visualizarse en caso de síntomas que indiquen la sospecha de un cáncer renal, como la presencia de sangre en la orina, el dolor en el costado, la fiebre o la detección de una prominencia en el abdomen en un examen médico rutinario. No obstante, alrededor del 30-50% de los pacientes presentan ya metástasis cuando son diagnosticados1,2, pues el cáncer renal tiende a producir metástasis enseguida, especialmente hacia los pulmones, y por tanto presentan un mal pronóstico, con una supervivencia media de sólo 6–8 meses.
Estos hechos hacen imprescindible la búsqueda de marcadores de daño renal que proporcionen la posibilidad de un diagnóstico temprano de la enfermedad. Hasta el momento se han estudiado diversos candidatos, tanto bioquímicos como moleculares, como ferritina, MN/CA9, alfafetoproteína, NMP (proteínas de la matriz nuclear), glucoproteínas asociadas a tumores (CEA, CA 50, CA 19.9, CA 125, CA 15.3), gammaenolasa, piruvato cinasa tipo M2, CD44, CD95, el gen supresor de tumores p53 e inestabilidad cromosómica, entre otros, pero ninguno de ellos ha resultado ser un marcador ideal, ya que, entre otras razones, no son lo suficientemente específicos3,4,.
El descubrimiento de las alteraciones genéticas que conducen a una célula renal al crecimiento incontrolado y entender los procesos que lo desencadenan son la base de las investigaciones dirigidas a la búsqueda de mejores marcadores5 que servirán para el desarrollo de estrategias terapéuticas más novedosas y eficaces.
En este artículo se realiza una revisión de los últimos avances en el conocimiento de las alteraciones observadas a escala molecular en cada tipo de RCC, así como en sus metástasis y la relación que estas alteraciones puedan tener con un mejor o peor pronóstico. También se revisan las posibilidades que hay en la actualidad para el tratamiento de la enfermedad, así como las líneas de investigación existentes hacia nuevas estrategias terapéuticas que van surgiendo con relación a los avances en el conocimiento de los sucesos moleculares que tienen lugar en el desarrollo del carcinoma renal.
Clasificación del carcinoma de células renalesSegún acuerdo internacional, la clasificación del carcinoma de células epiteliales renales debe hacerse no sólo en función de su procedencia histológica, sino también atendiendo a las alteraciones moleculares de base6,7. Así, según su morfología, su origen celular y las alteraciones genéticas, se distinguen 5 tipos de RCC:
- 1.
Tumores considerados malignos:
- –
Carcinoma de células claras: la célula de origen se localiza en el túbulo contorneado proximal de la nefrona y representa el 60-62% de los RCC. Es el cáncer renal convencional.
- –
RCC papilar: se origina en el túbulo contorneado distal. Tiene una incidencia de un 15% del total de RCC.
- –
RCC cromófobo: se origina en las células intercalares del túbulo colector. Son el 5-10% de los casos.
- –
Carcinoma del túbulo colector: se presenta sólo en menos del 1% de los casos.
- –
- 2.
Tumores considerados benignos. Oncocitoma: procede de células intercalares y representa el 7-10% del total.
Esta clasificación también tiene relevancia pronóstica. Así, por ejemplo, el pronóstico de pacientes con RCC cromófobo es significativamente mejor que el de aquellos que tienen RCC de células claras; entre los RCC papilares existe un subtipo, el 1, de células pequeñas y citoplasma escaso, que tiene un pronóstico mejor que el subtipo 2, cuyas células son grandes y con citoplasma eosinófilo8-10. En este sentido, hay que atender también al estadio del tumor, sea cual sea el tipo del que se trate. Para llevar a cabo la clasificación del estadío del tumor, el sistema más aceptado actualmente es el TNM Staging System11, que se introdujo por primera vez en 1978 y posteriormente ha sido modificado varias veces, hasta el modelo que hoy por hay se acepta como válido, que es el aceptado en 1997 por la American Joint Committee on Cancer (AJCC) y la International Union Against Cancer (UICC). Este sistema clasifica el tumor basándose en su tamaño, los nódulos linfáticos afectados y la invasión del tumor a la vena renal o incluso a la vena cava. Igualmente, tiene en cuenta el desarrollo de metástasis y su extensión12. Todos estos criterios de clasificación, según el estadio y dependiendo del tipo de tumor, proporcionan en conjunto una idea aproximada del pronóstico para el paciente afectado de cáncer renal.
Genética molecular y patogenia de los distintos tipos de carcinomas de células renalesEl hecho de que algunas formas del cáncer renal tengan un carácter hereditario proporciona la evidencia de que hay influencia genética en el desarrollo de la enfermedad. Los avances en metodología para el análisis cromosómico y del ADN han permitido detectar alteraciones genéticas asociadas a los tipos mayoritarios de carcinoma renal (tabla 1). El mapeo cromosómico y otras técnicas citogenéticas como el cariotipado espectral (SKY, que permite la visualización simultánea de todos los cromosomas en colores distintos para cada par y que permite detectar puntos de rotura cromosómica y visualizar traslocaciones fácilmente)13,14, la hibridación genómica comparativa (CGH, útil para detectar ganancias o pérdidas en el material cromosómico)13,15 o el análisis comparativo de micro-arrays de ADN (CGMA)16 aplicadas al análisis de tumores sólidos renales han permitido detectar y localizar diversos genes supresores de tumores y oncogenes implicados en la patogenia del RCC.
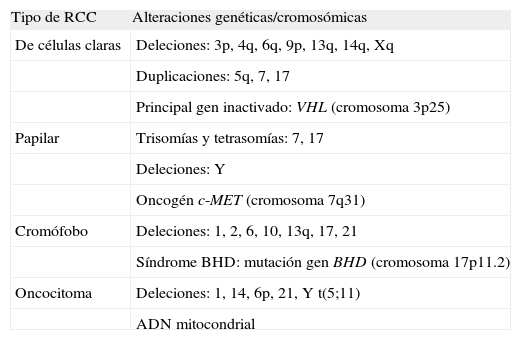
Alteraciones moleculares más frecuentes según el tipo de RCC
| Tipo de RCC | Alteraciones genéticas/cromosómicas |
| De células claras | Deleciones: 3p, 4q, 6q, 9p, 13q, 14q, Xq |
| Duplicaciones: 5q, 7, 17 | |
| Principal gen inactivado: VHL (cromosoma 3p25) | |
| Papilar | Trisomías y tetrasomías: 7, 17 |
| Deleciones: Y | |
| Oncogén c-MET (cromosoma 7q31) | |
| Cromófobo | Deleciones: 1, 2, 6, 10, 13q, 17, 21 |
| Síndrome BHD: mutación gen BHD (cromosoma 17p11.2) | |
| Oncocitoma | Deleciones: 1, 14, 6p, 21, Y t(5;11) |
| ADN mitocondrial |
BHD: Birt Hogg-Dubé; RCC: carcinoma de células renales; VHL: Von Hippel-Lindau.
Dentro de este tipo de RCC existen formas que se presentan de manera hereditaria, como en el síndrome de Von Hippel-Lindau (VHL), caracterizado por la presencia de carcinoma de células claras renales, hemangiomas, feocromocitomas, etc., o el carcinoma de células claras renales hereditario (HCRCC) y formas que se presentan de manera esporádica.
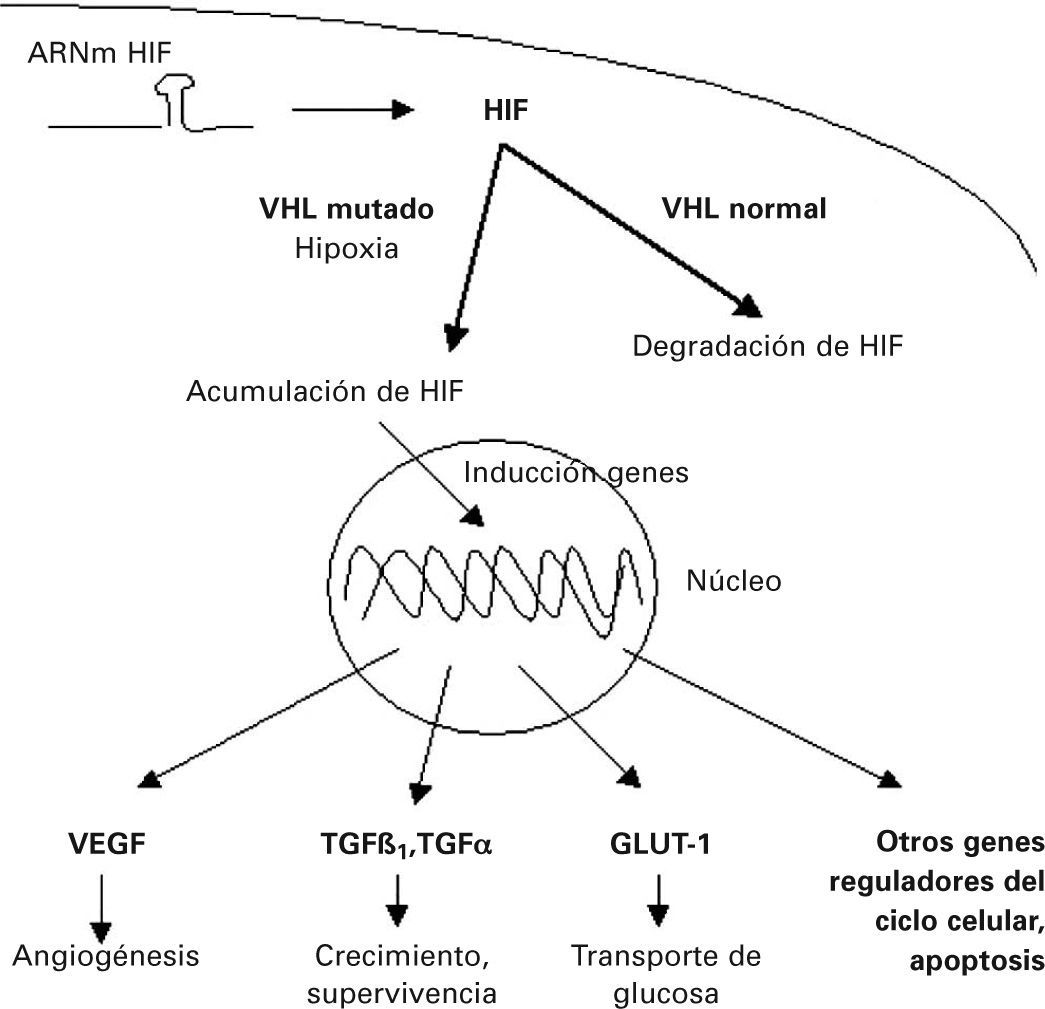
Al comparar el tejido tumoral y el tejido normal mediante CGH, las alteraciones cromosómicas observadas mayoritariamente implican pérdidas de material genético en los cromosomas 3p, 4q, 6q, 9p, 13q, 14q y Xq, y ganancias en los cromosomas 5q, 7 y 1717,18. De todas estas, las alteraciones que se han encontrado con más frecuencia son las deleciones en 3p. Estudios genéticos de pacientes con síndrome de Von Hippel-Lindau mostraron que todos ellos tenían inactivado un gen común, el gen VHL, localizado en el cromosoma 3p2519,20. Parece ser que el estado de este gen contribuye directamente a la regulación postraduccional del factor inducible de hipoxia (HIF). En situación fisiológica normal, el gen VHL contribuye a la degradación de HIF. En cambio, cuando hay un defecto o falta dicho gen, el factor HIF se acumula e induce la transcripción de genes sensibles a la hipoxia, como factores de la angiogénesis (VEGF), factores de crecimiento y supervivencia celular (TGF&1, TGFa), apoptosis, metabolismo de la glucosa (transportador de glucosa1) y otros genes reguladores del ciclo celular (fig. 1). Se considera un gen supresor de tumores, que también se encuentra inactivo en la mayoría de casos esporádicos de RCC de células claras21-24.
 por parte del gen VHL. GLUT-1: transportador de glucosa 1; TGF: factor de crecimiento tumoral; VEGF: factor de crecimiento endotelial vascular.")
Siguiendo el modelo clásico de los genes supresores de tumores, ambas copias deben estar mutadas para que se desencadene el proceso de tumoración. El mecanismo por el que se inactivan las 2 copias se ha visto que es distinto en las formas hereditarias y esporádicas. Los pacientes con enfermedad de VHL heredan una de las dos copias del gen mutado. Todas las células de su organismo son, pues, haploinsuficientes para dicho gen. En un segundo suceso, y esta vez en una única célula renal, sufrirían pérdida de la copia normal del gen (pérdida de heterocigosis [LOH]), por ejemplo, por una traslocación y pérdida del gen en la siguiente división celular, conservando sólo la copia anormal y, por tanto, sufriendo inactivación de VHL25,26. En cambio, en caso de cáncer renal de células claras esporádico, el primer suceso ocurre ya en una única célula del riñón, por mutación adquirida en uno de los alelos del gen, dando lugar a la pérdida de éste (LOH). Un segundo suceso en la misma célula hemicigota (por traslocación, metilación, mutación puntual o deleción) daría lugar a la pérdida de actividad del alelo normal y, por tanto, al genotipo tumoral26-28. No obstante, la pérdida de VHL es sólo el inicio del proceso tumoral.
Por otra parte, se encuentran los cánceres renales de células claras hereditarios distintos de VHL (HCRCC), en los que se ha visto que poseen una traslocación constitucional que implica también al cromosoma 3, pero no al gen VHL. Dicha translocación facilitaría la pérdida de parte del cromosoma 3 en una célula, por ejemplo renal, y crearía la predisposición al desarrollo del tumor por aumento de la probabilidad de sufrir una segunda alteración en el cromosoma 3 normal26.
La gran heterogeneidad genética observada en tumores renales que en principio parecen histológicamente iguales hace difícil establecer una línea clara de sucesos genéticos que definan el proceso de tumorogénesis29. Así, entre los cambios observados en cáncer renal de células claras está el aumento de actividad de genes promotores del crecimiento (oncogenes como c-fos, c-myc), pérdida de genes que inducen la muerte celular (bcl-2), genes promotores de la angiogénesis (VEGF, PDGF) y muchos otros30-32. Todos estos genes serían candidatos para el uso de terapias antitumorales.
Estudios más recientes se inclinan por la búsqueda de marcadores tempranos de lesión tubular renal, que permitan detectar que una célula renal posee alteraciones genéticas que la van a destinar al crecimiento incontrolado, pero antes de que se haya producido el tumor. Mediante la técnica RAP-PCR y comparando tejido renal normal y tumoral, uno de los genes que se detectó sobreexpresado en células tumorales fue el hHAVcr-1 (receptor celular 1 humano del virus de la hepatitis A), que se sitúa en el cromosoma 5q (cromosoma que se ha visto duplicado en el 60% de los carcinomas de células claras). Además se ha visto que la sobreexpresión de este gen es específica del epitelio tubular proximal y, aunque la función del producto para el que codifica no está clara en células renales normales, no sólo parece estar implicado en la tumorogénesis (desdiferenciación celular, invasión y metástasis), sino que también ha sido encontrado en orina de pacientes con necrosis tubular aguda (ATN). Este hecho hace pensar que la detección de esta proteína en orina puede ser un marcador importante de daño tubular en el diagnóstico temprano de carcinoma de células claras. Los ensayos llevados a cabo en monos proporcionan resultados esperanzadores en el uso de terapias que tienen como objetivo esta proteína, pues se ha visto que las inmunotoxinas contra el homólogo de hhavcr-1 producen la destrucción de células renales que sobreexpresan dicha proteína33.
Carcinoma papilar de células renalesLos análisis citogenéticos y moleculares de este tipo de tumores muestran trisomías y tetrasomías de los cromosomas 7 y 17, y pérdidas del cromosoma Y como las alteraciones más frecuentes34.
En familias con cáncer papilar renal hereditario se ha visto como mutación más común la del protooncogén c-MET, dando lugar al correspondiente oncogén. MET se encuentra en la región cromosómica 7q31 y codifica para la proteína c-met, receptor celular del factor de crecimiento de hepatocitos (HGF). El receptor mutado es incapaz de inactivarse tras la unión de HGF, lo que conduce a la activación de diversos transductores de señal intracelulares, provocando una respuesta en la célula que la lleva al crecimiento incontrolado e invasivo35,36. Al tratarse de un oncogén con sólo un alelo mutado, ya se obtiene el fenotipo tumoral, a diferencia de lo que ocurre en el ccRCC, en el que son necesarias varias alteraciones sucesivas.
Pero no todos los pacientes con cáncer renal papilar tienen mutado el gen c-MET. En el caso de tumores esporádicos de este tipo, se ha visto mutación de c-MET en sólo un 13% de los casos; en cambio, sí se ha comprobado ganancia del cromosoma 7, lo que puede indicar que, aunque el receptor no esté mutado, una sobreexpresión de éste es suficiente para la tumorogénesis, pues la respuesta celular a HGF aumentaría37,38.
En este sentido, y dependiendo de la cantidad de material genético duplicado observado en el tumor, especialmente en 7p y 17p, que se desarrolla típicamente en pacientes con enfermedad renal quística o pacientes que necesitan hemodiálisis, los tumores papilares se han podido diferenciar en 2 subtipos, con diferente pronóstico. También se ha relacionado la mutación en el gen de la fumarato hidratasa con una predisposición a RCC papilar de tipo 239; por otra parte, la pérdida del cromosoma Xp se asocia a un tipo de tumor papilar fulminante40.
Otras alteraciones genéticas que se han observado en algunos tumores papilares son la sobreexpresión de genes de factores de crecimiento endotelial vascular, factores de la angiogénesis, y oncogenes como c-myc y c-fos, lo mismo que ocurre en muchos carcinomas de células claras, lo que indicaría que, aunque ambos tipos de cáncer renal surjan a partir de alteraciones genéticas diferentes, podrían converger en los mismos defectos a lo largo de su evolución41.
Carcinoma cromófobo de células renalesLos RCC cromófobos están considerados cánceres malignos, pero presentan un pronóstico mejor que los dos tipos anteriormente descritos. Sin embargo, también pueden evolucionar a metástasis, especialmente hepática, dependiendo de las alteraciones genéticas que se presenten. Las alteraciones genéticas observadas en este tipo de cáncer renal son numerosas: se han hallado pérdidas en los cromosomas 1, 2, 6, 10, 13q, 17 y 21, dando lugar a un genotipo casi haploide42, y en tumores que han desarrollado metástasis se han encontrado alterados, además, los cromosomas 5, 12, 15 y 1843. Estos hallazgos hacen difícil definir un suceso concreto y único que desencadene la formación de este tipo de tumores.
Existe una enfermedad hereditaria, el denominado síndrome BHD (Birt Hogg-Dubé), en la que, entre otras alteraciones, se presenta predisposición a desarrollar tumor renal de tipo cromófobo. Esta enfermedad es autosómica dominante, lo que indica que este tipo de tumores, al igual que los dos anteriores, puede presentarse tanto de forma esporádica como hereditaria. El estudio de las alteraciones genéticas que se presentan en este síndrome ha conducido a la identificación de un nuevo gen implicado en el desarrollo de cáncer renal, localizado en el cromosoma 17p11.2, que muestra en este tipo de síndrome una alta frecuencia de inserciones/deleciones. Este gen codifica para una proteína denominada foliculina, que se expresa en los tejidos afectados en dicha enfermedad (renal, pulmonar y dermis), pero cuya función en éstos todavía no está clara44.
OncocitomaLos oncocitomas se desarrollan a partir de las células intercalares del túbulo colector renal, igual que el RCC cromófobo, pero son diferentes histológicamente, y se consideran tumores benignos, pues en la mayoría de los casos son asintomáticos y no necesitan cirugía.
Se conoce muy poco acerca de las alteraciones genéticas que los producen. Se han visto, entre otros, alteraciones en el ADN mitocondrial45 o traslocación recíproca entre los cromosomas 5 y 11. En un estudio realizado mediante la técnica CGH se observó que 6 de los 13 tumores examinados presentaban pérdidas en el cromosoma 1 y/o en el 14. Otras alteraciones observadas incluían pérdidas en los cromosomas 6p, 21 e Y. Las pérdidas de los cromosomas 1 y/o 14, zonas en las que se encuentran codificados genes que regulan el crecimiento celular, podrían ser las alteraciones genéticas iniciales para el desarrollo de los oncocitomas46.
Alteraciones genéticas en la metástasis del carcinoma de células renalesAl examinar las alteraciones genéticas presentes en tumores de metástasis de RCC y comparar los hallazgos obtenidos con las alteraciones que presentaba el tumor primario, se ha visto que los primeros difieren sustancialmente del tumor renal de origen. El estudio por CGH indica que las alteraciones encontradas más comúnmente en tumores de metástasis implican a los cromosomas 3p, 4q, 6q, 8p y 9p, en cuanto a pérdidas de material genético se refiere, y a 17q y Xq, en cuanto a ganancias. También se encontró alta amplificación genética en la zona 11q22-2347.
Pero no sólo se han visto diferencias entre el tumor metastásico y el primario, sino que también hay diferencias entre las propias metástasis, es decir, el número de ganancias o pérdidas de material genético encontradas en tumores extendidos a nódulos linfáticos o a pulmón es menor que el encontrado en otros órganos afectados, lo que indica que la diseminación del tumor por la sangre es fruto de la adquisición de alteraciones genéticas más complejas. Podría ser que las diferentes metástasis deriven de clones del tumor primario distintos o, lo que es más probable, que el tumor primario sufra sólo unas determinadas alteraciones genéticas que le den ventaja para la invasión a otros tejidos y, una vez ocurrido esto, el resto de alteraciones más complejas observadas en las distintas metástasis se desarrollarían en la evolución de la propia metástasis47.
Alteraciones genéticas y pronósticoEl pronóstico del paciente afectado de cáncer renal no sólo depende de la tipificación histológica del tumor y el estadio en el que se encuentre. Las alteraciones genéticas que se presentan en la célula tumoral desempeñan un papel fundamental en la evolución del tumor y, por lo tanto, su estudio debe tenerse en cuenta para determinar la mayor o menor gravedad del cáncer.
Moch et al48 realizaron un estudio en el que se demostró que existe relación entre el número total de alteraciones genéticas en el tumor y un peor pronóstico. Sin embargo, cuando se analizaron por separado las ganancias y las pérdidas de material genético, se vio que sólo las pérdidas presentaban una asociación significativa con un peor pronóstico. La explicación residiría en que la pérdida de la expresión de numerosos genes supresores de tumores por deleción cromosómica probablemente facilita la progresión del tumor. Cuando se analizó la asociación entre el pronóstico y el número de amplificaciones o deleciones presentes en cada cromosoma por separado, se encontró, por ejemplo, que la pérdida del cromosoma 9p era el único evento relacionado con la recurrencia del tumor. Es probable que en esta zona exista un gen supresor de tumores cuya pérdida esté implicada en la progresión del tumor.
Por otra parte, en ocasiones el carcinoma renal puede transformarse en sarcoma, que es muy agresivo y presenta un pronóstico mucho peor. Los análisis por CGH de tumores sarcomatoides de RCC mostraron que los sarcomas son genéticamente mucho más complejos en cuanto a las alteraciones que presentan49. Las zonas delecionadas más prevalentes en este tipo de formaciones se encontraron en 13q y 4q, y las duplicaciones más frecuentes, en 17, 7 y 8q. Además también se detectó una alta tasa de expresión de los loci 11q22-23 y 7p21-22 que no se presentaba en el área del tumor adyacente no sarcomatosa, lo que indica que en estos loci podría existir un oncogén implicado en la transformación sarcomatosa.
Posibilidades terapéuticas actuales y futurasHoy por hoy, las opciones terapéuticas (tabla 2) existentes para el RCC sólo son efectivas cuando la enfermedad se detecta a tiempo y no se ha desarrollado metástasis. Así, en los casos en que el tumor está localizado en un sólo riñón, el tratamiento de elección es la cirugía, procediéndose a la extracción quirúrgica completa del riñón y de los posibles ganglios linfáticos afectados.
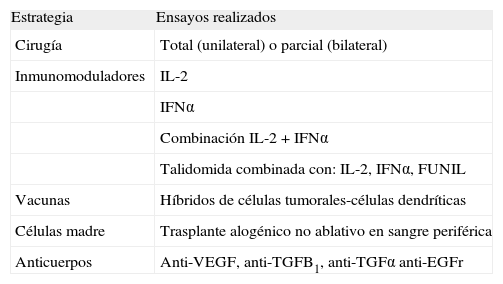
Posibilidades terapéuticas para el cáncer renal actuales y en experimentación
| Estrategia | Ensayos realizados |
| Cirugía | Total (unilateral) o parcial (bilateral) |
| Inmunomoduladores | IL-2 |
| IFNα | |
| Combinación IL-2 + IFNα | |
| Talidomida combinada con: IL-2, IFNα, FUNIL | |
| Vacunas | Híbridos de células tumorales-células dendríticas |
| Células madre | Trasplante alogénico no ablativo en sangre periférica |
| Anticuerpos | Anti-VEGF, anti-TGFB1, anti-TGFα anti-EGFr |
EGFr: receptor del factor de crecimiento epitelial; FUNIL: IL-2 + IFNα + 5-fluorouracilo; IFNα: interferón a; IL-2: interleucina 2; TGF: factor de crecimiento tumoral; VEGF: factor de crecimiento endotelial vascular.
Pero existen pacientes, como los afectados de VHL, en los que el tumor es bilateral, y en tal caso la nefrectomía debe ser parcial. En los casos en que el tumor ha invadido la vena renal o la vena cava pero todavía no se ha propagado hacia otros órganos, la cirugía aún puede ofrecer una buena probabilidad de curación. No ocurre así cuando se desarrolla metástasis, que además en este tipo de cáncer es muy temprano. El pronóstico en estos casos es malo: la cirugía no es efectiva, y tampoco lo son la radiación y los anticancerígenos tradicionales (quimioterapia). En estos casos, el tratamiento más corriente es con interleucina 2, único fármaco cuyo uso ha sido aprobado por la Food and Drug Administration para el tratamiento de esta enfermedad. Con la administración de este fármaco se trata de estimular la capacidad del sistema inmunitario para destruir el cáncer.
Otra sustancia de similar mecanismo de acción ensayada ha sido el interferón alfa, con respuestas similares a la anterior, favorecidas por factores como el tipo de tumor del que se trate (grado de agresividad), nefrectomía previa (se ha visto que la esperanza de vida media aumenta en un 50% en comparación con los que reciben sólo interferón50) o metástasis limitadas sólo a los pulmones. Como para conseguir una tasa de respuesta más o menos alta se necesita la administración de altas dosis, y esto es perjudicial por la gran incidencia de efectos secundarios, se han llevado a cabo estudios para definir estrategias que permitan disminuir la dosis y obtener una respuesta similar. Se comparó la respuesta de los pacientes tratados con una dosis alta de interleucina 2, una dosis alta de interferón alfa y la combinación de ambos fármacos a dosis más bajas, obteniéndose respuestas positivas (reducción de la metástasis) en el 17, el 15 y el 18,6% de los casos, respectivamente. No obstante, aunque la combinación de ambos fármacos produjo una mayor respuesta, se vio que el índice de toxicidad del tratamiento también era mayor, y además no se producía un aumento significativo en la esperanza de vida media51.
Recientemente se ha introducido en este tipo de ensayos otro agente inmunomodulador, la talidomida, con propiedades antiangiogénicas y que parece ser más potente que el interferón (IFN) a o la IL-2. El ensayo de terapias combinadas, talidomida con IFNa o con IL-2 o con ambas y además 5-fluorouracilo (FUNIL), ha demostrado gran eficacia en numerosos ensayos clínicos realizados en pacientes en el estadio temprano de la enfermedad, en los que se observa que no progresa el tumor52.
No obstante, los bajos índices de respuesta obtenidos con los tratamientos actuales para la metástasis de RCC y el hecho de que la única vía de curación para el cáncer renal localizado sea la nefrectomía completa hacen necesaria la búsqueda de nuevas estrategias terapéuticas. Con el mayor conocimiento actual del proceso tumorogénico renal se abren nuevos caminos de investigación.
Las investigaciones actuales están encaminadas al descubrimiento de terapias más específicas contra el RCC que no presenten toxicidad. Dado que el poder antigénico del tumor renal ha sido demostrado con el uso de citocinas, se han llevado a cabo ensayos con terapias novedosas que tratan de inducir una respuesta más potente del sistema inmunitario contra el cáncer53. Así, Kugler et al54 desarrollaron una vacuna de híbridos de células tumorales-células dendríticas que, ensayada en 17 pacientes, tuvo resultados bastante esperanzadores: 4 de ellos tuvieron remisión completa del cáncer, en 2 remitió parcialmente, en otros 2 la enfermedad permaneció estable y en el resto no hubo respuesta significativa y la enfermedad siguió progresando. La ventaja de este tipo de terapia es que no presentó toxicidad.
Otra estrategia ensayada de tipo inmunoterapéutico ha sido el trasplante de células madre alogénico no ablativo en sangre periférica, que se realizó en 19 pacientes con metástasis renal, de los que la mitad presentó regresión de la enfermedad, y en 3 de ellos el cáncer remitió de forma completa55. No obstante, todavía no hay conclusiones definitivas acerca del uso de este tipo de terapias.
Otras investigaciones van encaminadas al uso de terapias inhibidoras de la angiogénesis para evitar la propagación del cáncer, pues los pacientes de RCC expresan gran cantidad de factores angiogénicos como VEGF, TGF^1 o TGFa. El uso de anticuerpos que neutralizan la acción de estos factores ha demostrado inhibición de la progresión del tumor21,56. Asimismo, el uso de antagonistas del receptor de EGF también ha causado regresión del RCC en modelos animales57.
El desarrollo de terapias similares en humanos, junto con el desarrollo de terapias génicas que consiguieran reemplazar la función de VHL (causante de la mayoría de los ccRCC) o bloquear el oncogén c-MET (causante del carcinoma papilar de riñón), podría proporcionar grandes beneficios en la lucha contra esta enfermedad.
ConclusiónGracias a los últimos avances en técnicas citogenéticas y de biología molecular, se ha podido conocer muchas de las alteraciones celulares causantes de la tumorogénesis de los distintos tipos de cáncer renal. Ahora se conoce, por ejemplo, que la mayoría de los ccRCC se desarrollan en asociación con la inactivación de ambas copias del gen VHL o que el carcinoma papilar renal está ligado a la mutación o sobreexpresión del gen c-MET, y que alteraciones de un gen localizado en el cromosoma 17 podrían ser la causa del carcinoma de tipo cromófobo.
Asimismo, el examen histológico para conocer el tipo de tumor del que se trata y su estadio, relacionado con las alteraciones genéticas presentes, puede proporcionar una idea aproximada del pronóstico del paciente.
Los avances en el conocimiento de las vías implicadas en el desarrollo del RCC permiten a los investigadores la búsqueda de nuevas posibilidades terapéuticas menos tóxicas, más específicas y, por tanto, más eficaces que las existentes en la actualidad. De todos modos, aunque se conocen algunos marcadores relacionados con la patogénesis del cáncer, como VEGF, TGFa o TGF^1, entre otros, son necesarios más estudios para el descubrimiento de otros genes supresores de tumores/oncogenes implicados, definir sus interacciones con otros factores reguladores del ciclo celular y la secuencia en que se produce su desregulación y que define el fenotipo tumoral y su evolución clínica, para obtener terapias cada vez mejores y aplicables a cada caso.
Además, debido a la precocidad del desarrollo de metástasis de este tipo de cáncer, se hace de vital importancia la búsqueda de marcadores tempranos fácilmente detectables de daño tubular renal, pues se podría evitar el desarrollo del tumor a tiempo y aumentar así la esperanza de vida de las personas afectadas o con riesgo de padecer esta enfermedad.
