




La enfermedad renal es un problema de salud pública por su incidencia, prevalencia y morbimortalidad. La inflamación y fibrosis son el motor de toda alteración del daño renal, independientemente de su origen. El proceso inflamatorio y fibrótico se caracteriza por infiltración de células inflamatorias, liberación de citoquinas, activación de fibroblastos, de señales químicas y vías de señalización. Este proceso conlleva la generación de células efectoras productoras de matriz extracelular o de complejos de ataque que desemboca en el daño orgánico. Ninguna terapia ha demostrado eficacia total frente al daño inflamatorio o fibrótico. Muchos fármacos, en desarrollo experimental, ejercen su inmunomodulación a través de la regulación de la actividad de muchos tipos de células inmunes y presentan un perfil antiinflamatorio y fibrótico. El objetivo es ser conocedores de la importancia de los procesos que son capaces de provocar el proceso inflamatorio y fibrótico dado que podría tratarse de marcadores diagnósticos y dianas terapéuticas.
Chronic kidney disease is a serious public health problem, due to its high incidence and prevalence, as well as its significant morbidity and mortality. Inflammation and fibrosis are the final step in renal failure. The inflammatory and fibrotic process is highlighted by infiltration by inflammatory cells, cytokine release, fibroblast accumulation, and activation of numerous chemical signals. Those processes involve the generating of immunomodulatory cells that produce an extracellular matrix and attack complex, leading to organ damage. There is no an effective therapy against fibrotic and inflammatory damage. There are different drugs that have shown to be beneficial over inflammation and fibrosis in experimental and in vitro studies. The aim is to be aware of the processes that are able to trigger fibrosis and inflammation, given that they could be used as diagnostic markers and therapeutic targets.
La nefrona es la unidad funcional del riñón; se compone del corpúsculo renal (glomérulo y cápsula de Bowman), la red tubular, el intersticio, aparato yuxtaglomerular y un entretejido vascular. El glomérulo se encarga de la formación de un ultrafiltrado a partir del plasma que pasa por los capilares glomerulares, la membrana basal y los podocitos con su hendidura de filtración compuesta por un complejo de diversas proteínas altamente especializadas, como la nefrina, esenciales en la permeabilidad glomerular1. El entramado túbulo-intersticial representa el 80% del volumen renal y se compone de1:
- •
Células intersticiales tipo I: fibroblastos like encargadas de la síntesis y degradación de matriz extracelular (MEC).
- •
Células intersticiales tipo II: monocitos derivados de macrófagos y células dendríticas presentadoras de antígenos.
- •
MEC: colágeno, proteinoglicanos, glicoproteínas, fluido intersticial.
- •
Células tubulares: encargadas del transporte de sustancias.
- •
Células especializadas: productoras de renina, vitamina D activa, eritropoyetina, Klotho.
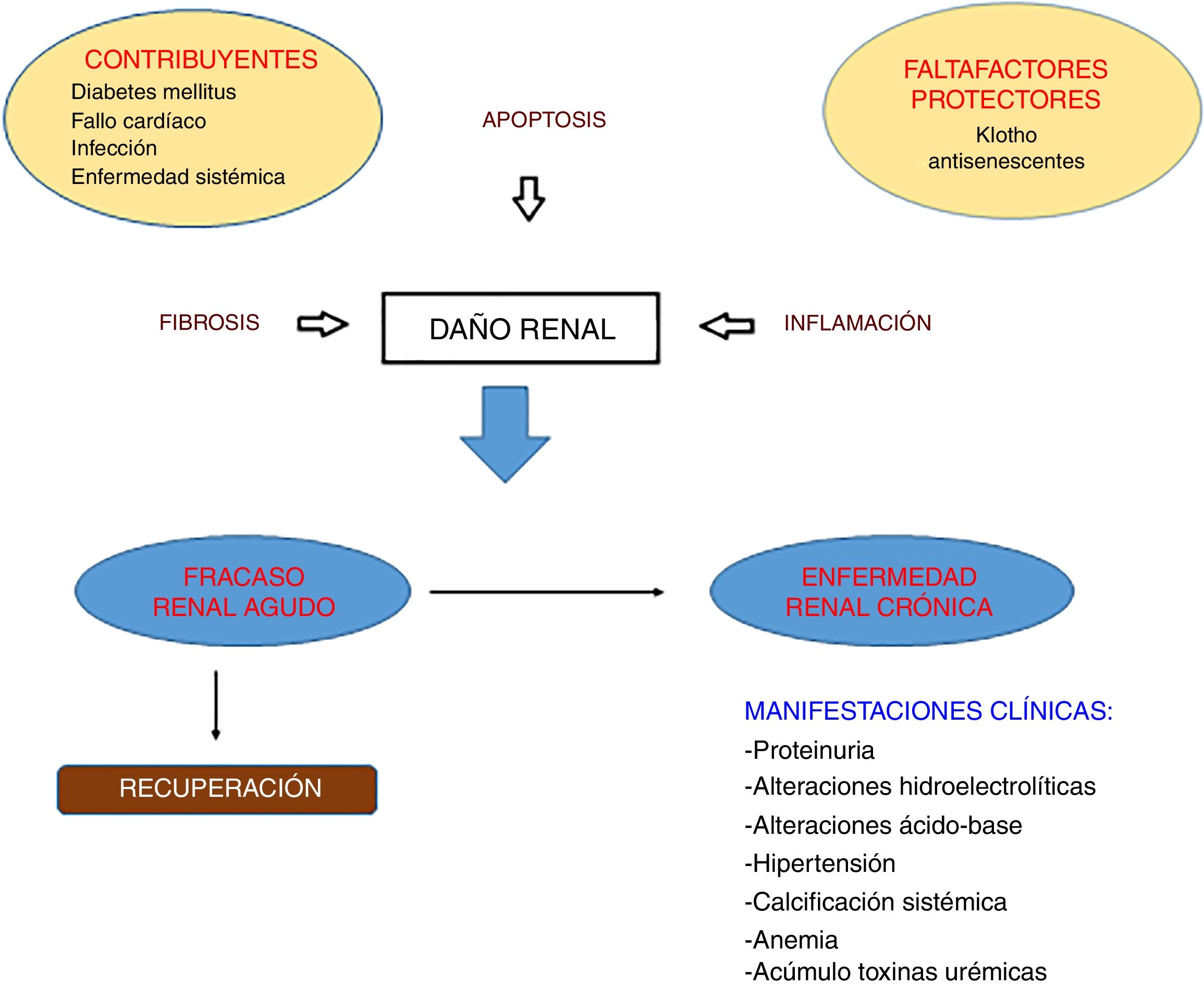
Ante una agresión se produce una respuesta reparadora inflamatoria que, si se perpetúa en el tiempo, deriva en una respuesta fibrótica conllevando el daño y el fallo orgánico. El tejido renal sufrirá una serie de eventos: activación de células residentes con capacidad de secreción de citoquinas y quimioquinas proinflamatorias, aumento de la permeabilidad vascular, rarefacción vascular, generación de MEC y el proceso de transición epitelio mesenquimal (TEM)2–6.
El riñón lesionado es incapaz de depurar estas sustancias proinflamatorias perpetuándose el daño; a eso hemos de añadir las comorbilidades del paciente con enfermedad renal crónica (ERC) o aguda y las de las técnicas de sustitución renal (tabla 1) generándose una alteración estructural y funcional permanente. Convendría conocer los pasos del daño tisular para así obtener métodos diagnósticos precoces y dianas terapéuticas que reviertan el proceso.
Causas y manteniemiento de la inflamación en la enfermedad renal
| Enfermedad renal | Hemodiálisis | Diálisis peritoneal |
|---|---|---|
| Disminución del aclaramiento de citoquinas | Infección del acceso vascular | Peritonitis |
| Acumulación de PGA, citoquinas etc. | Bioincompatibilidad diálisis | Bioincompatibilidad diálisis |
| Comorbilidades | Exposición a endotoxinas del dializado | Exposición a endotoxinas del dializado |
| Comorbilidades | Comorbilidades |
PGA: productos de glicación avanzada.
La agresión renal, independientemente de su etiología o que sea aguda o crónica, tienen nexos en común derivados de su fisiopatología. Este vínculo es el proceso inflamatorio y fibrótico, ambos relacionados con la mortalidad en el fallo renal. La inflamación es un proceso tisular constituido por fenómenos moleculares, celulares y vasculares con finalidad defensiva frente a agresiones; conlleva la liberación de mediadores bioquímicos con diversas funciones y finalmente la reparación3,4. Si esta respuesta inflamatoria se mantiene conllevará la respuesta fibrótica consistente en la formación excesiva de tejido conectivo como consecuencia de un proceso reparativo, reactivo o inflamatorio crónico3,4. Los hallazgos anatomopatológicos de la fibrosis renal se caracterizan por glomeruloesclerosis, fibrosis tubulointersticial, infiltración inflamatoria, y la pérdida de parénquima renal caracterizado por atrofia tubular, pérdida capilar y agotamiento podocitario.
Las células con mayor relevancia en el proceso de fibrosis son el fibroblasto y el miofibroblasto. Ambas proceden del tejido conectivo; el miofibroblasto ha desarrollado algunas características de las células musculares lisas que pueden adaptar marcadores de otras células mesenquimales3,7. Secretan proteínas, colágeno, procolágeno, tropoelastina y proteoglicanos para formar parte de la MEC7 y presentan receptores para estrógenos, esteroides, hormona tiroidea, vitamina D, factores del crecimiento, y a nivel renal, podrían expresar eritropoyetina7. Su origen, en el proceso fibrótico, es diverso y ha sido objeto de debate en la literatura científica; así LeBleu et al.8, en un modelo de fibrosis renal en ratones manipulados genéticamente, demostraron las diversas fuentes de los miofibroblastos: el 50% provenían de fibroblastos residentes, un 35% de células madre mesenquimales pluripotenciales de la médula ósea y tan solo un 15% del proceso de TEM o de la transdiferenciación endotelio-mesenquimal. Estudios como el de Zeisberg et al.9 o Inoue et al.10 dan más relevancia al origen del fibroblastos y miofibroblastos a la TEM; este proceso consiste en que células epiteliales sufren, bajo circunstancias proinflamatorias, una transformación a células mesenquimales (ej. fibroblastos) adquiriendo sus marcadores bioquímicos9,10 (α−SMA [actina del músculo liso], vimentina, CD31, colágeno I, fibronectina), sus funciones y aumentan la produccción de MEC; también pierden sus marcadores epiteliales9,10 (E-Cadherina, Zonula ocludens-1). Otras posibles fuentes de miofibroblastos son los pericitos vasculares11,12 y, como ya hemos mecionado, la médula ósea13,14.
Otro evento destacable en el daño renal es la pérdida de microvasculatura renal y su densidad que se correlaciona con la severidad de fibrosis en los pacientes con ERC y fallo renal agudo6 (FRA). Esta alteración conlleva hipoxia tisular que desencadena una respuesta inflamatoria y fibrótica15–17. La hipoxia estimularía la producción de factores proangiogénicos como el factor de crecimiento endotelial vascular18 (VEGF), o el factor inducido por hipoxia el cual en situaciones de hipoxemia evita su prolil hidroxilación y promovería la angiogénesis pero también es conocido que vía factor de crecimiento transformante β (TGF- β) promueve la expansión de la MEC en el intersticio y la degeneración vascular19,20. Presumiblemente el daño capilar comienza con una alteración de la mitocondria endotelial con imposibilidad de generar ATP15 lo que conlleva la pérdida de uniones entre las células endoteliales y finalmente su apoptosis21 y necrosis; también la pérdida de sirtuína podría influir al dificultarse la angiogénesis22, y de factores de supervivencia endotelial como VEGF. Como consecuencia se aumenta la permeabilidad capilar y el edema intersticial. Todo ello conllevaría isquemia tisular que agrava más la degeneración endotelial, el daño microvascular6,15 y la respuesta inflamatoria y fibrótica6,15,17,18.
No hemos de olvidar mencionar el proceso de apoptosis celular y su papel en el daño renal. La apoptosis promueve la pérdida celular tanto en el FRA como en la ERC, pero también presenta un efecto beneficioso dado que evita las respuestas proliferativas celulares exacerbadas23. En las zonas de lesión compiten factores de supervivencia celular (factor de crecimiento hepático, ciclina, nefrina) con factores letales (Bad, Bax) expresados por las propias células residentes u otro tipo de células que son atraídas ante la lesión21,23. Pero el proceso apoptoico se podría perpetuar al promoverse las señales proapoptoicas e inflamatorias con una exacerbación del daño tisular. Señalar que la apoptosis puede generar necrosis si las células apoptoicas no son eliminadas o bien ante la ausencia de generar energía21.
Lo que diferencia un proceso reparativo «sano» y el proceso inflamatorio-fibrótico sigue estando a debate en la comunidad científica. Una diferencia importante es la duración del daño: una lesión aguda provoca una reacción inflamatoria y activación de fibroblastos similar a la hallada en la ERC. Pero, a diferencia del fallo crónico, en el fallo agudo se produce una reparación debida a la regeneración tubular y el remodelado de la matriz. Si el daño se prolonga el tejido reacciona de una manera anómala con la sobreproducción de MEC. Una posible explicación podría ser que en el daño crónico la cascada de eventos mencionados se ve amplificada. Las consecuencias clínicas del proceso de inflamación y fibrosis son: a nivel glomerular el paso de proteínas a la red tubular (proteinuria) generando toxicidad celular3,10; a nivel tubulointersticial puede provocar alteraciones del manejo hidroelectrolítico y de determinados solutos (fig. 1).
Factores relevantes en la inflamación y la fibrosis
Distintas citoquinas, factores de crecimiento, y partículas con efecto inflamatorio y fibrótico se han visto implicados en el desarrollo de la enfermedad renal. A continuación resumiremos brevemente los más relevantes:
- •
Proteasas24: enzimas secretadas por monocitos y macrófagos. Digieren componentes de la membrana basal glomerular alterando su permeabilidad y provocando hematuria.
- •
Sistema del complemento25: sistema proteico constituido por 26 elementos. Su activación se produce por la vía alterna (por lipopolisacáridos bacterianos), la vía clásica (por complejos inmunes, endotoxinas y ARN de algunos virus) o por la vía de las lectinas. Este sistema participa en la eliminación de complejos inmunes y modulando la respuesta humoral y celular. Algunos de sus fragmentos son mediadores inflamatorios (C3b y C5a); a nivel renal el denominado complejo de ataque de membrana (C5-C9) puede producir lesión lítica cuando la formación de complejos inmunes ocurre sobre la membrana celular; o lesión sublítica donde se producen trastornos en el funcionamiento normal de la célula, que incluyen síntesis de citocinas y mediadores que la célula normalmente no produce. Tiene relevancia en la patogenia de glomerulopatía C3 o en microangiopatía trombótica.
- •
Radicales libres de oxígeno26 (ROS): moléculas químicamente inestables producidas en pequeñas cantidades por el organismo; son muy reactivas produciendo oxidación celular, alteraciones en el ADN y cambios que aceleran el envejecimiento. Su producción se debe a distintas células infiltrantes: polimorfonucleares y macrófagos. Estimulan el NF-κB (nuclear factor-κBeta) y con ello la cascada inflamatoria con la producción de citocinas y quimiocinas. Alteran la permeabilidad y potencian la degradación de la membrana basal glomerular. A su vez provocan alteraciones en la hemodinámica renal con reducción del filtrado glomerular (FG).
- •
Factores de coagulación: en determinadas entidades de daño renal (microangiopatía trombótica, glomerulopatía rápidamente progresiva, lupus eritematoso sistémico) se ha objetivado un disbalance entre el sistema de coagulación y el sistema fibrinolítico27; de hecho en biopsias renales se ha comprobado la presencia de fibrina. Al haber disfunción endotelial se activa la cascada de la coagulación: el factor Von Willebrand favorecería la adhesión plaquetaria y la formación de coágulos. Además diferentes estirpes celulares podrían liberar citocinas con efecto procoagulante27. Mención especial merece el factor de crecimiento derivado de plaquetas (PDGF); a nivel renal participa en el reclutamiento y proliferación de células mesenquimales y podría tratarse de un biomarcador de determinadas patologías renales como la nefropatía IgA28.
- •
Eicosanoides29: compuestos derivados de ácidos grasos que actúan como mediadores locales. Son sintetizados por diferentes estirpes celulares (macrófagos, plaquetas, células residentes activadas). Engloban prostaglandinas, leucotrienos, tromboxanos y lipoxinas; sus efectos incluyen: proliferación de células mesangiales y endoteliales, aumento en la producción de MEC y moléculas de adhesión, aumento en la expresión del complejo mayor de histocompatibilidad, efecto proinflamatorio y quimiotáctico.
- •
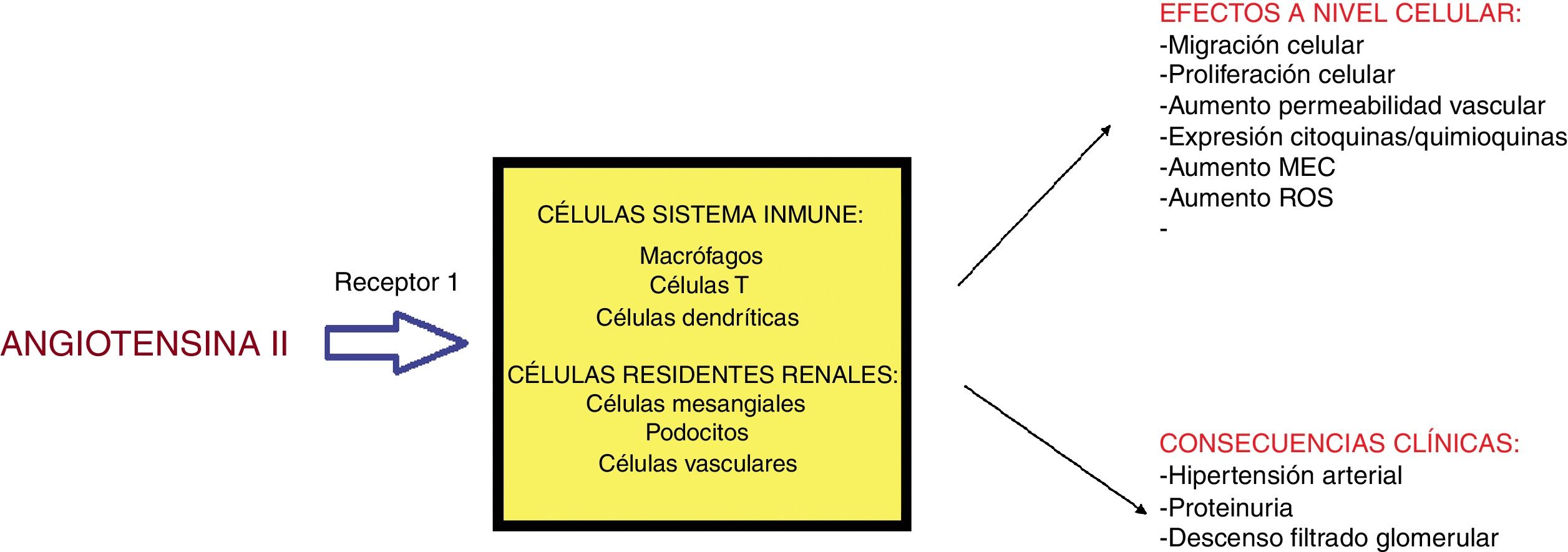
Angiotensina II (A-II): hormona peptídica derivada del angiotensinógeno perteneciente al sistema renina-angiotensina-aldosterona (RAAS). El estímulo de renina conlleva la síntesis de A-II en el pulmón; esta actúa a través de 2 receptores: tipo 1 (efecto vasoconstrictor, proinflamatorio y fibrótico) y el tipo 2 con efectos antagónicos30. Sus acciones incluyen: estímulo en la síntesis de la hormona antidiurética a nivel hipofisario, de la sed, del sistema nervioso simpático y la absorción de sodio y agua en el túbulo proximal; los efectos hemodinámicos de la A-II conllevan vasoconstricción periférica30; este mismo efecto vasoconstrictor se produce sobre la arteriola eferente renal en un intento de mejorar el FG pero a la larga puede provocar la vasoconstricción de la arteriola aferente con el efecto contrario30. El riñón presenta un «sistema RAAS local» con efectos proinflamatorios, proliferativos y profibróticos actuando sobre distintas estirpes celulares (fig. 2). Así, la A-II promueve la activación de ROS, la migración celular, el remodelado de la matriz y puede activar señales intracelulares que conllevan daño tisular31. También favorece el reclutamiento de células inflamatorias al inducir producción de quimioquinas30. La mayoría de estos efectos son a través de su receptor tipo I. La aldosterona presenta funciones proinflamatorias similares29. Su papel en el desarrollo de proteinuria (nociva y tóxica celular) sería el siguiente: en el fallo renal, al disminuir el número de nefronas, se produce hipertensión glomerular aumentando la presión hidrostática mediado, en parte, por la A-II. Este estrés mecánico incrementa los receptores tipo 1 y aumenta la producción de A-II en los podocitos. La pérdida del contacto directo entre los podocitos y la mayor permeabilidad a la albúmina provoca la fuga de esta a la luz tubular30. Además, A-II disminuye la expresión de nefrina o podocina glomerular perpetuándose el paso de proteínas a la red tubular.
- •
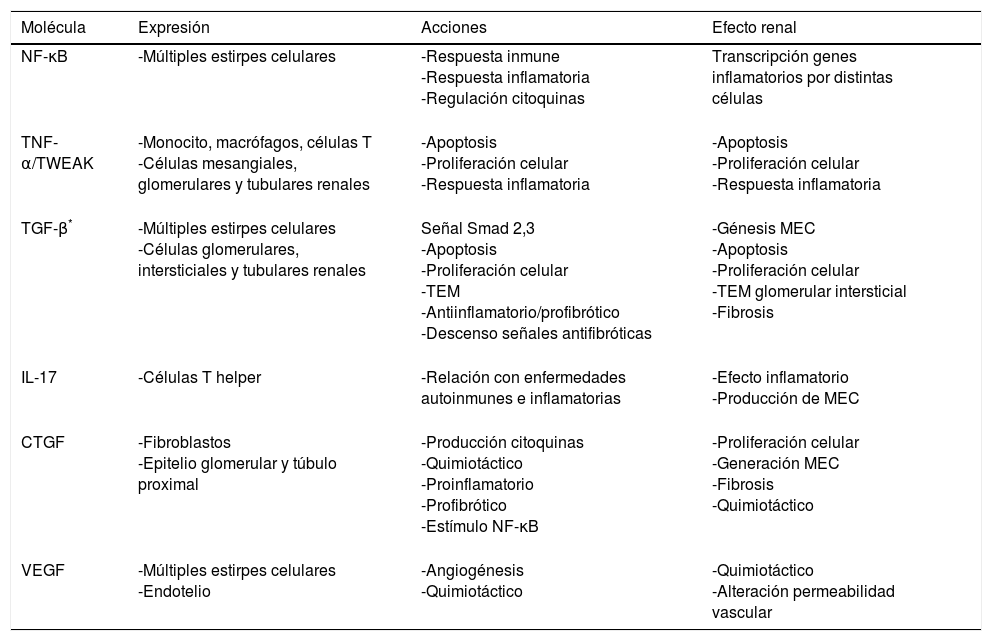
Citocinas y quimiocinas: proteínas sintetizadas por distintas estirpes celulares. Presentan efectos paracrinos, yuxtacrinos y autocrinos regulando la inflamación y la respuesta inmune4. Pueden estimular células mesangiales, epiteliales y fibroblastos provocando un cambio en ellas que conlleva el acúmulo de MEC4,10. A continuación mencionaremos algunas de las más relevantes (tabla 2).
Tabla 2.Esquema de las acciones de diferentes citoquinas con papel relevante en la injuria renal
Molécula Expresión Acciones Efecto renal NF-κB
-Múltiples estirpes celulares
-Respuesta inmune
-Respuesta inflamatoria
-Regulación citoquinas
Transcripción genes inflamatorios por distintas células TNF-α/TWEAK -Monocito, macrófagos, células T
-Células mesangiales, glomerulares y tubulares renales
-Apoptosis
-Proliferación celular
-Respuesta inflamatoria-Apoptosis
-Proliferación celular
-Respuesta inflamatoriaTGF-β*
-Múltiples estirpes celulares
-Células glomerulares, intersticiales y tubulares renalesSeñal Smad 2,3
-Apoptosis
-Proliferación celular
-TEM
-Antiinflamatorio/profibrótico
-Descenso señales antifibróticas
-Génesis MEC
-Apoptosis
-Proliferación celular
-TEM glomerular intersticial
-FibrosisIL-17 -Células T helper -Relación con enfermedades autoinmunes e inflamatorias
-Efecto inflamatorio
-Producción de MECCTGF -Fibroblastos
-Epitelio glomerular y túbulo proximal-Producción citoquinas
-Quimiotáctico
-Proinflamatorio
-Profibrótico
-Estímulo NF-κB
-Proliferación celular
-Generación MEC
-Fibrosis
-QuimiotácticoVEGF -Múltiples estirpes celulares
-Endotelio-Angiogénesis
-Quimiotáctico-Quimiotáctico
-Alteración permeabilidad vascularMEC: matriz extracelular; TEM: transición epitelio mesenquimal.

- -
NF-κB32:. factor de transcripción, de distintas estirpes celulares, compuesto por proteínas diméricas que pertenecen a la familia Rel. En reposo se encuentra unido a su fracción ikB, que al fosforilarse sufre una translocación al núcleo. Numerosas zonas de ADN de genes inflamatorios, citoquinas, glucosa y virus tienen zonas de unión con NF-κB. Sus genes de transcripción tienen un papel relevante en la inflamación y respuesta inmune: proteína atrayente de monocitos (MCP-1), molécula de adhesión celular vascular 1 (VCAM-1), molécula de adhesión intercelular 1 (ICAM-1), interleucina (IL) 8 etc.
- -
Factor de necrosis tumoral α (TNF-α)33: producido por monocitos, macrófagos, células T, células mesangiales, glomerulares y tubulares renales como respuesta a un daño celular. Induce apoptosis, síntesis y secreción de otras citoquinas; es clave en la respuesta inflamatoria. TWEAK (TNF like weak inducer of apoptosis), está encargado de la apoptosis, proliferación y proceso inflamatorio en las células tubulares renales en presencia de citoquinas proinflamatorias.
- -
TGF-β: la superfamilia TGF-β consiste en péptidos secretados, donde las tres isoformas de TGF-β (TGF-β 1, 2, y 3), activinas y proteínas morfogénicas óseas (BMP) son las más estudiadas. TGF-β está expresado ampliamente a nivel celular y actúa, sobre todos los tipos de células debido a la participación de una cascada de señalización intracelular de proteínas de la familia Smad34. La señal de TGF-β es transducida a través de su receptor de membrana celular tipo I y tipo II serina/treonina quinasa; esto desencadena la fosforilación y la activación de Smad-2 y Smad-334. En el núcleo la señal Smad, unida al receptor, participa directamente en la activación transcripcional de los genes diana relacionados con el proceso de fibrosis34. A su vez TGF-β y Smad-7, sinérgicamente, inducen apoptosis in vitro34. Muchas de las acciones de TGF-β pueden ser independientes de la señal Smad, actuando a través de otras señales como la proteína RhoA (gen homólogo Ras, familia A), proteínas quinasas activadas por mitógeno (MAPK)35 etc. TGF-β se encuentra aumentado en ERC34,35; se considera un importante regulador de la proliferación, diferenciación y apoptosis celular, respuesta inmune y generación de MEC; también posee acciones antiinflamatorias2,34. Además es esencial en el proceso de TEM2,34 y en la activación de miofibroblastos sobre distintas estirpes celulares36, así como de otras vías de señalización que perpetúan el daño renal (tabla 3). La expresión de TGF-β está regulada al alza por leptina, distintas citoquinas, IL-1 (aumenta sus acciones no su expresión)34 y es inhibido por vitamina D y sus análogos, celecoxib, BMP-7, el factor de crecimiento hepático entre otros34.
Tabla 3.Efectos de TGF−β sobre el riñón
Glomérulo Apoptosis podocitaria:
-Deplección podocitaria
-Adhesión penacho celular
-Menor supervivencia cel. endotelialApoptosis cel. endotelial:
-Menor supervivencia célula endotelial
-Pérdida capilar glomerularActivación cel mesangiales:
-Acúmulo MECGlomeruloesclerosis
TGF β Túbulo
intersticioApoptosis cel. epitelial:
-Degeneración tubular
-Atrofia tubularTEM:
-Degeneración tubular
-Activación fibroblastosActivación fibroblastos:
-Aumento MECFibrosis tubulointersticial Degeneración tubular:
-Menor supervivencia cel. endotelial
-Atrofia tubular
-Pérdida capilar tubular - -
Células T helper 17 (CTH17)/IL-17 y otras IL: las CTH17 representan un subconjunto de células que secretan IL-17 A y F, IL-6 e IL-22; siendo IL-17 la que presenta mayor efecto inflamatorio y promueve la síntesis de colágeno en distintos modelos de fibrosis, incluido el renal37,38. Las CTH17 se han relacionado con diversas enfermedades autoinmunes e inflamatorias crónicas39. Hay un equilibrio entre las células CTH17 y las células T reguladoras que depende del factor de transcripción del ácido retinoico relacionado al receptor γt, Stat 3 y 5 (transducción de señal y activador transcripción 3 y 5) y el factor de transcripción FoxP3. De esta manera se regula la respuesta inmune mediante la secreción de citoquinas pro y antiinflamatorias37. Las citoquinas principales que regulan el balance de las CTH17 y células T reguladoras son TGF-β e IL-638. Las células T reguladoras, descendidas en la ERC, son esenciales en prevenir la acumulación de fibrocitos y su bloqueo podría aumentar el acúmulo de fibrosis y el daño orgánico40. Otras IL destacables son la IL-1 e IL-8 con efecto quimiotáctico, IL-6 podría actuar como factor de crecimiento celular y proinflamatorio29.
- -
Factor de crecimiento del tejido conectivo41 (CTGF): proteína rica en cisteína aumentada en los estados profibróticos. Eleva otras citoquinas y quimioquinas: MCP-1, RANTES (citoquina expresada y secretada por el linfocito T normal), IL-6, interferón α, y reduce señales antiinflamatorias como la IL-10. Potencia la actividad del factor NF-κB y es factor de atracción para monocitos, macrófagos y células T en la zona tubulointersticial renal.
- -
VEGF6,42: regulador de la óxido nítrico sintetasa endotelial. Se ha relacionado con las alteraciones podocitarias y endoteliales en la nefropatía diabética. Los niveles de VEGF en la ERC son variables: disminuidos se asocian con deficiencia de la angiogénesis y apoptosis celular y aumentados con el reclutamiento de macrófagos en ratones con déficit de óxido nítrico sintetasa.
- •
Leptina/adiponectina43: adiponectina es una proteína que aumenta la sensibilidad a la insulina y posee acciones antiinflamatorias y antiaterogénicas. La leptina se considera proaterogénica y fibrogénica, estimula a las células endoteliales renales y produce un aumento de la producción de TGF-β y de colágeno tipo IV. Ambas aumentan en la ERC por disminución de su aclaramiento.
- •
Nanopartículas calcioproteicas: su acúmulo en la ERC se debe al disbalance del metabolismo fosfocálcico que se origina en la enfermedad óseo-mineral. Su internalización por células macrófagos-like provoca una respuesta proinflamatoria44.
- •
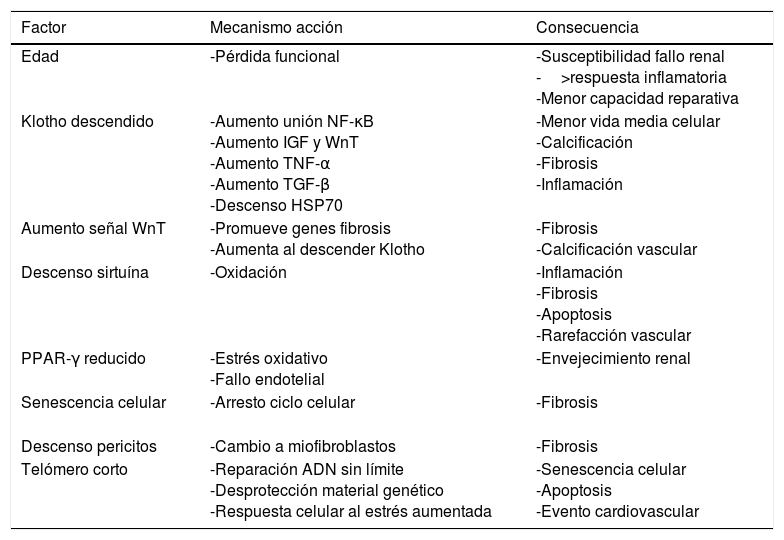
Envejecimiento renal: el riñón añoso sufre una serie de cambios estructurales que trastocan la hemodinámica, el comportamiento fisiológico renal y su respuesta frente a las agresiones. Estos cambios perjudican la capacidad renal para recuperarse de una lesión con mayor susceptibilidad al fallo renal, a una mala recuperación y tendencia a la progresión de la enfermedad45. Hasta el 73% de los pacientes mayores de 70 años donantes renales presentan algún grado de esclerosis45. Diversos mecanismos son los que hacen proclive al fallo renal al riñón añoso por métodos relacionados con la inflamación y fibrosis46 (tabla 4): descenso de Klotho, aumento de la señal WnT, descenso de sirtuína, descenso de PPARγ, descenso de pericitos a favor de fibroblastos, telómero corto etc.
Tabla 4.Mecanismo de lesión renal por riñón añoso
Factor Mecanismo acción Consecuencia Edad -Pérdida funcional -Susceptibilidad fallo renal
->respuesta inflamatoria
-Menor capacidad reparativaKlotho descendido -Aumento unión NF-κB
-Aumento IGF y WnT
-Aumento TNF-α
-Aumento TGF-β
-Descenso HSP70-Menor vida media celular
-Calcificación
-Fibrosis
-InflamaciónAumento señal WnT -Promueve genes fibrosis
-Aumenta al descender Klotho-Fibrosis
-Calcificación vascularDescenso sirtuína -Oxidación -Inflamación
-Fibrosis
-Apoptosis
-Rarefacción vascularPPAR-γ reducido -Estrés oxidativo
-Fallo endotelial-Envejecimiento renal Senescencia celular
-Arresto ciclo celular -Fibrosis Descenso pericitos -Cambio a miofibroblastos -Fibrosis Telómero corto -Reparación ADN sin límite
-Desprotección material genético
-Respuesta celular al estrés aumentada-Senescencia celular
-Apoptosis
-Evento cardiovascular - •
Otros42,47: MCP-1 (recluta monocitos y macrófagos en el proceso de inflamación renal), productos de glicación avanzada, B-catenina (inhibe nefrina provocando proteinuria masiva2), oncostatina M, acúmulo y síntesis renal de lípidos (expresión de TGF-β), inhibidor de plasminógeno, quininas e histaminas (efectos vasomotores y de alteración en la permeabilidad vascular); micro-ARN (presentan capacidad de regular la expresión de otros genes; los hay negativos frente a la fibrosis); vía de señalización Janus kinase/Signaling transducers and activation of transcription, regula la expresión génica de mediadores proinflamatorios, de diferenciación celular y activación TGF-β, producción de colágeno tipo IV y fibronectina; vía Snail1 (promotor de TEM).
El diagnóstico de la enfermedad renal viene determinado por una historia y anamnesis detallada. Los parámetros bioquímicos clásicos renales (creatinina, FG, proteinuria) nos aportan una idea de la cuantía del daño renal así como su clasificación en grados. Si se sospecha un mecanismo inmunológico relacionado con enfermedades sistémicas deberemos de solicitar parámetros asociados como autoanticuerpos: ANA, anticuerpos anti-DNA, ANCA, inmunoglobulinas, complemento etc. Muchas veces es prioritario un diagnóstico temprano de la disfunción renal; los marcadores clásicos, como creatinina (influenciada por el estado nutricional, peso, sexo etc.), fallan en este sentido, sin embargo los denominados «nuevos biomarcadores» (NGAL, KIM 1, LFABP etc.) están ganando cada día más terreno especialmente en el FRA48.
Muchas de las moléculas mencionadas anteriormente servirían para aproximarnos al proceso que acontece en el riñón; especialmente las relacionadas con la fibrosis. La determinación de CTGF urinario se postula como un biomarcador que nos daría una idea del proceso de fibrosis a nivel renal41; otro ejemplo sería el receptor del factor de crecimiento epidérmico y el propio factor (EGF)49. Este desempeña un papel importante en la regeneración tubular durante el FRA con reducción de sus niveles. Por otro lado, la expresión de receptor del factor de crecimiento epidérmico es mayor en condiciones como nefropatía crónica de injerto y glomerulonefritis con medias lunas, lo que sugiere que ligandos distintos a EGF, como CTGF, pueden activar la señalización por este receptor; constituye por lo tanto un objetivo terapéutico en los casos de ERC pero no en el FRA.
Para determinar de una manera certera qué es lo que está ocurriendo a nivel celular en el daño renal tendríamos la práctica de la biopsia renal.
Medidas terapéuticas frente a la fibrosis y progresión de fallo renalNo existe un arsenal terapéutico eficaz al 100% en el fallo renal. En este apartado queremos profundizar sobre medidas terapéuticas dirigidas específicamente frente al evento inflamatorio y fibrótico o eventos relacionados con la progresión de la injuria renal.
- -
Terapia frente a la apoptosis: actuar sobre la vía de las caspasas podría ser una alternativa en la injuria renal. El uso del inhibidor de las caspasas IDN-6556ha disminuido la lisis hepatocitaria en la hepatitis C50. También pifitrina, inhibidor de p53 (supresor tumoral), protege a las células del daño sobre el ADN que provocan agentes de quimio y radioterapia y es nefroprotector en modelos de isquemia-reperfusión y cisplatino51. Otras medidas: uso de inhibidores de Apaf-1 (factor activador de apoptosis)52.
- -
Terapia frente al infiltrado celular: el acúmulo celular intersticial se debe a la interacción entre quimiocinas y receptores de membrana de los leucocitos53. Una vez producida esa interacción los leucocitos producirían gran cantidad de sustancias proinflamatorias y profibróticas42. El actuar sobre receptores de quimioquinas podría evitar el reclutamiento celular. El bloqueo de CCR1, 2 y 5 (C-C chemokine receptor type 1, 2, 5), esenciales en la migración leucocitaria intersticial y la infiltración glomerular54, ha tenido un impacto significativo en el acúmulo leucocitario, fibrosis intersticial, lesión tubular y mejoría de la función renal en modelos animales55. También se han desarrollado terapias frente a quimiocinas como MCP-1: emapticap pegol que lo neutraliza56. Otro fármaco que actúa en decremento del acúmulo de estirpes celulares sería micofenolato mofetilo (inhibe la síntesis de purinas al inhibir la enzima inosin monofosfato deshidrogenasa) que disminuye la proliferación de linfocitos T57. Micofenolato se ha empleado con éxito en nefropatía IgA, membranosa, lúpica y en trasplante renal57.
- -
Terapia frente a citoquinas: entre las posibles terapias cuyo target son citoquinas proinflamatorias o fibróticas encontramos la inhibición de PDGF con trapidil que provocaría un descenso en la proliferación y la acumulación de MEC58. Otra arma terapéutica sería el bloqueo de determinadas IL proinflamatorias como la IL-2 mediante el uso de anticuerpos monoclonales (basiliximab) especialmente indicado en trasplante renal59. La inhibición de NF-κB podría resultar interesante; no obstante, su inhibición total podría ser perjudicial60. De manera parcial puede ser inhibido con inhibidores de la enzima conversora de angiotensina, estatinas, inhibidores de la calcineurina, LF15-019560. Otro bloqueo importante sería el de TGF-β bien con anticuerpos como 1D11 o SB-50512461 (reducen los niveles de Smad 2, 3), Bendavia15 (protector de la mitocondria tubular) o con determinadas moléculas (Klotho o BMP-7) como veremos posteriormente.
- -
Antisenescentes: una de las moléculas con un futuro prometedor en evitar la progresión de la disfunción renal es Klotho62 (síntesis predominantemente renal). Posee efectos antioxidantes, antisenescentes, antiinflamatorios, antifibróticos (inhibe TGF-β) y anticalcificantes62–64. Zhao et al.63 demostraron que Klotho podría ser un modulador de la inflamación, actuando negativamente sobre NF-κB en un modelo de ratones diabéticos; y Liu et al.64 comprobaron que suprime el gen 1 de ácido retinoico con lo que se disminuía la expresión de IL-6 e IL-18 in vitro e in vivo. En modelos experimentales de FRA, se ha demostrado el descenso de Klotho y, por tanto, su papel como posible biomarcador65. Su reposición podría conllevar la recuperación del daño renal65.
- -
Moléculas con efecto antiinflamatorio o fibrótico: el uso de IL-4, IL-3, IL-10 podrían mitigar los efectos inflamatorios en la patología renal en modelos experimentales66. Otros como los PPARγ inhiben la diferenciación de CTH17 y descienden la producción de IL-1766. Los inhibidores COX-2 son moduladores negativos de la cascada inflamatoria con menor respuesta fibrótica66. La pentoxifilina tiene un efecto frente a CTGF, MCP-1, TNF-α y TGF-β.66 Los inhibidores de mTOR (de amplio uso en trasplante renal) podrían tener un papel relevante como antifibróticos, habiendo demostrado un descenso de la inflamación y de la fibrosis en modelos experimentales de nefropatía membranosa67. Mención aparte supone el efecto de BMP-7; de síntesis predominantemente renal regula negativamente el proceso fibrótico en distintos modelos de enfermedad renal, hepática o cardíaca66,68. En general se mantiene un equilibrio entre TGF-β y BMP-7 en el control del proceso fibrótico68; ambas moléculas se unen a receptores tipo I serintreonina kinasa que desencadenan diferentes señales intracelulares mediadas por las proteínas Smad. La adición de BMP-7 exógeno bloquearía la señal TGF-β aminorándose el proceso fibrótico68.
- -
Terapia frente a la rarefacción vascular: ya comentamos la relación que hay entre la pérdida de densidad capilar y el deterioro de la función renal6,15–17. Diferentes estudios han investigado el uso de medidas a la hora de mitigar este evento que además podrían tener otros efectos beneficiosos. Tal es el caso de Bendavia (protector mitocondrial); así Liu et al.15 sobre un modelo de fallo renal isquémico en ratas demostró que el tratamiento con Bendavia promovía la angiogénesis con una reducción de la rarefacción vascular, además presentaba propiedades antifibróticas y antiinflamatorias (descenso TGF-β y TNF-α) con mejoría de la función renal. Este mismo grupo ya había demostrado la protección de este fármaco sobre células tubulares69. Las células estromales adiposas también han resultado ser una excelente terapia en la mejora de la rarefacción vascular en modelo de fallo renal animal isquémico70. Estas células además de poseer efectos antiinflamatorios, antiapoptópicos, presentan efectos proangiogénicos. En el estudio de Collet et al.70 se demostró una reducción de la respuesta inflamatoria y aumento de la actividad vascular y de su densidad. También se ha empleado el uso de factores de crecimiento como VEGF71, de stem cells endoteliales72 o la activación de sirtuína22.
- -
Fármacos o técnicas empleados en la enfermedad renal que podrían influir sobre el estado inflamatorio:
- 1)
Inhibidores RAAS: el uso de inhibidores de la enzima conversora de angiotensina, antagonistas de receptores de A-II, y los inhibidores de la aldosterona presentan un efecto antiproteinúrico demostrado73. No obstante el efecto nefroprotector de los inhibidores del eje RAAS van más allá; provocan un descenso en la proliferación y fibrosis dependiente de citocinas asociadas a la activación de NF-κB y, en consecuencia, disminución de la progresión de daño renal74. Si tenemos en cuenta todos los efectos nocivos de A-II (ya expuestos anteriormente) es de prever que su inhibición es beneficiosa en la progresión del fallo renal al aminorar el evento inflamatorio y fribrótico31,74.
- 2)
Vitamina D/análogos: tienen un papel esencial en el tratamiento del hiperparatiroidismo secundario. Pero los efectos pleitrópicos de vitamina D activa han abierto una estrategia terapéutica prometedora. En modelos experimentales de lesión renal vitamina D/análogos han demostrado75,76 inhibir el gen de la renina y NF-κB; inhibir la infiltración inflamatoria y fibrótica; regresar la TEM y el acúmulo de MEC; inhibir RANTES, TNF-α, TGF-β, NF−κ≡ y otras citoquinas; incrementar el receptor para endotelina B; disminuir la proteinuria. En distintos modelos de fibrosis se ha demostrado que la expresión del receptor de vitamina D queda abolido casi por completo77 por lo que su suplementación podría implicar mejora del daño renal y de la fibrosis. Tan et al.77, en un modelo animal de uropatía obstructiva e in vitro en células tubulares humanas, comprobaron una reducción de los depósitos de fibronectina y colágeno e inbición de la señal TGF-β tras la adición de paricalcitol (análogo de vitamina D). No obstante muchos de estos efectos son a nivel experimetal animal o in vitro sin modelo humano comprobado.
- 3)
Captores del fósforo66: sevelámero presenta acciones de mejoría del perfil lipídico, descenso de parámetros inflamatorios, descenso de factor de crecimiento fibroblástico 23 (FGF-23) y aumento de Klotho.
- 4)
Diálisis: la biocompatibilidad en diálisis ha conseguido controlar la respuesta inflamatoria en los pacientes de diálisis peritoneal y hemodiálisis78. No obstante estos efectos no han sido estudiados a nivel del proceso de deterioro de la función renal, pero podría tratarse de un posible beneficio en el FRA o en mantener la función renal residual.
- -
Stem cells (SC): el riñón tiene cierta capacidad de regeneración celular. Existen SC a nivel glomerular y porción S3 del túbulo proximal con capacidad de regenerar podocitos79; no obstante la autorregeneración renal es baja y a veces puede ser errática79. También SC procedentes de la médula ósea se han hallado en zonas de lesión renales y son capaces de diferenciarse en células de estirpe renal80. Las SC que se transdiferencian son capaces de generar un microambiente apropiado con la secreción de determinadas sustancias/citocinas y factores que promueven la reparación renal aunque puedan tener también efectos deletéreos. Partiendo de esa base, el uso de SC podría resultar útil en la reparación del daño orgánico. En modelos animales de FRA el trasplante de SC mesenquimales procedentes de médula ósea injertaron con éxito en células epiteliales tubulares con mejoría de la función renal81. La fuente externa de SC podrían ser las células embrionarias (derivadas de los blastocitos) y las SC pluripotenciales66,81,82; no obstante el uso de células embrionarias no diferenciadas puede provocar teratomas. Las SC pluripotenciales (provenientes de células somáticas adultas) podrían ser una alternativa válida82.
El proceso de injuria renal es extremadamente complejo debido al proceso inflamatorio y fibrótico; no solo por la cantidad de eventos celulares y moleculares que tienen lugar sino porque el propio daño renal perpetúa esta cascada de señales proinflamatorias y fibróticas y disminuye las antagónicas de estos efectos. El conocimiento del proceso celular de daño renal debido a la inflamación y fibrosis representa una diana tanto diagnóstica como terapéutica.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
