


La granulomatosis con poliangeitis (GPA) o granulomatosis de Wegener es una vasculitis necrosante de afectación sistémica y causa desconocida, que afecta a vasos de mediano y pequeño calibre con compromiso principalmente renal y respiratorio, descrita por primera vez por Klinger en 1931 y posteriormente en 1936 por Friedrich Wegener1. La afectación de los tejidos blandos orbitarios por la enfermedad se estima en torno al 45-60%, siendo el 8-16% la forma de presentación inicial2. En estos casos los pacientes pueden presentar síntomas que van desde proptosis, diplopía o visión reducida, hasta simplemente edema palpebral, siendo en ocasiones, la única manifestación de la enfermedad3.
Para el diagnóstico de la GPA clásicamente se han utilizado una serie de criterios (American College of Rheumatology 19904):
- 1.
Inflamación nasal u oral: úlceras.
- 2.
Alteraciones de la radiografía de tórax: nódulos, cavitación o infiltrados no migratorios ni fugaces.
- 3.
Alteraciones en el sedimento urinario: microhematuria o cilindros hemáticos.
- 4.
Presencia de inflamación granulomatosa en la biopsia.
Dos de estos 4 criterios predicen la enfermedad con una sensibilidad del 88% y una especificidad del 92%.
El diagnóstico actual de la enfermedad se basa en la combinación de los criterios clínico-patológicos, la determinación de anticuerpos anticitoplasma de neutrófilos (ANCA) y la biopsia. La positividad para ANCA aparece en el 90% de los pacientes que presentan enfermedad en fase activa, mientras que baja hasta un 63% en pacientes con una enfermedad inactiva1. Solamente el 50-65% de las formas limitadas serán ANCA positivas2.
Los estudios más recientes hablan de incidencias en torno a 11,3 por millón y prevalencias de 145,9 por millón en el Reino Unido5, sin diferencias entre sexos y cierta predominancia en personas de raza blanca, aunque es probable que esta incidencia esté infraestimada a consecuencia de formas leves localizadas. Es una enfermedad rara en la infancia y presenta un pico de incidencia en la quinta década de la vida.
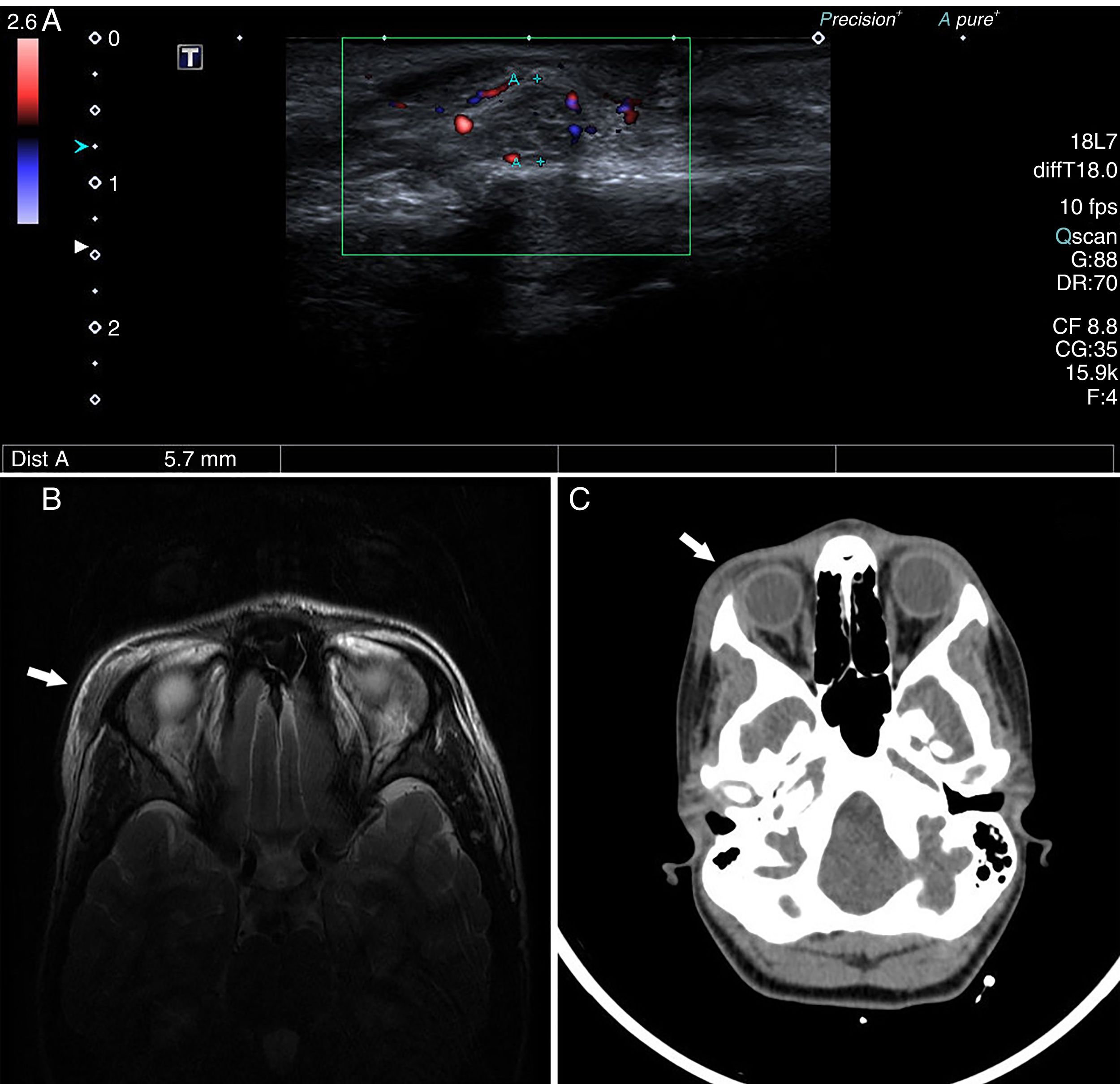
Presentamos el caso de una adolescente de 15 años que acudió a consultas de atención primaria pediátrica por edema palpebral superior derecho de mayor intensidad por las mañanas, sin otra sintomatología acompañante ni etiología evidente. Como antecedente presentó, 3 meses antes, mononucleosis infecciosa positiva para EBV y CMV. Tras un mes de evolución, debido a la ausencia de mejoría y a la aparición de un nódulo periorbitario, acudió de nuevo decidiéndose realizar una ecografía (fig. 1A) que resultó inespecífica, y aunque sugirió etiología inflamatoria, no se descartaron otras causas al observarse 2 tumoraciones en reloj de arena. Se amplió el estudio con RMN (fig. 1B) y TAC (fig. 1C), que mostraron la presencia de importante tumefacción de la musculatura orbicular en el reborde externo de la órbita derecha, así como 2 nódulos de apariencia inflamatoria sin poder descartar un carácter tumoral.
 Ecografía en la que se observa engrosamiento del músculo orbicular derecho a nivel supraciliar externo, con cambios inflamatorios en la grasa subyacente, observando 2 tumoraciones que presentan una morfología en reloj de arena de márgenes mal definidos e hipervasculares en el estudio Doppler. B) RMN que muestra tumefacción de la musculatura subcutánea orbicular, ligeramente hiperintensa con respecto a la musculatura normal y con ganancia moderada de contraste paramagnético tras la inyección endovenosa del mismo. C) TAC en la que se aprecia dicha tumefacción sin condicionar afectación ósea ni infiltración de las estructuras intraorbitarias. Dichos estudios sugieren origen inflamatorio sin poder descartar carácter tumoral.")
Estudio mediante pruebas de imagen. A) Ecografía en la que se observa engrosamiento del músculo orbicular derecho a nivel supraciliar externo, con cambios inflamatorios en la grasa subyacente, observando 2 tumoraciones que presentan una morfología en reloj de arena de márgenes mal definidos e hipervasculares en el estudio Doppler. B) RMN que muestra tumefacción de la musculatura subcutánea orbicular, ligeramente hiperintensa con respecto a la musculatura normal y con ganancia moderada de contraste paramagnético tras la inyección endovenosa del mismo. C) TAC en la que se aprecia dicha tumefacción sin condicionar afectación ósea ni infiltración de las estructuras intraorbitarias. Dichos estudios sugieren origen inflamatorio sin poder descartar carácter tumoral.
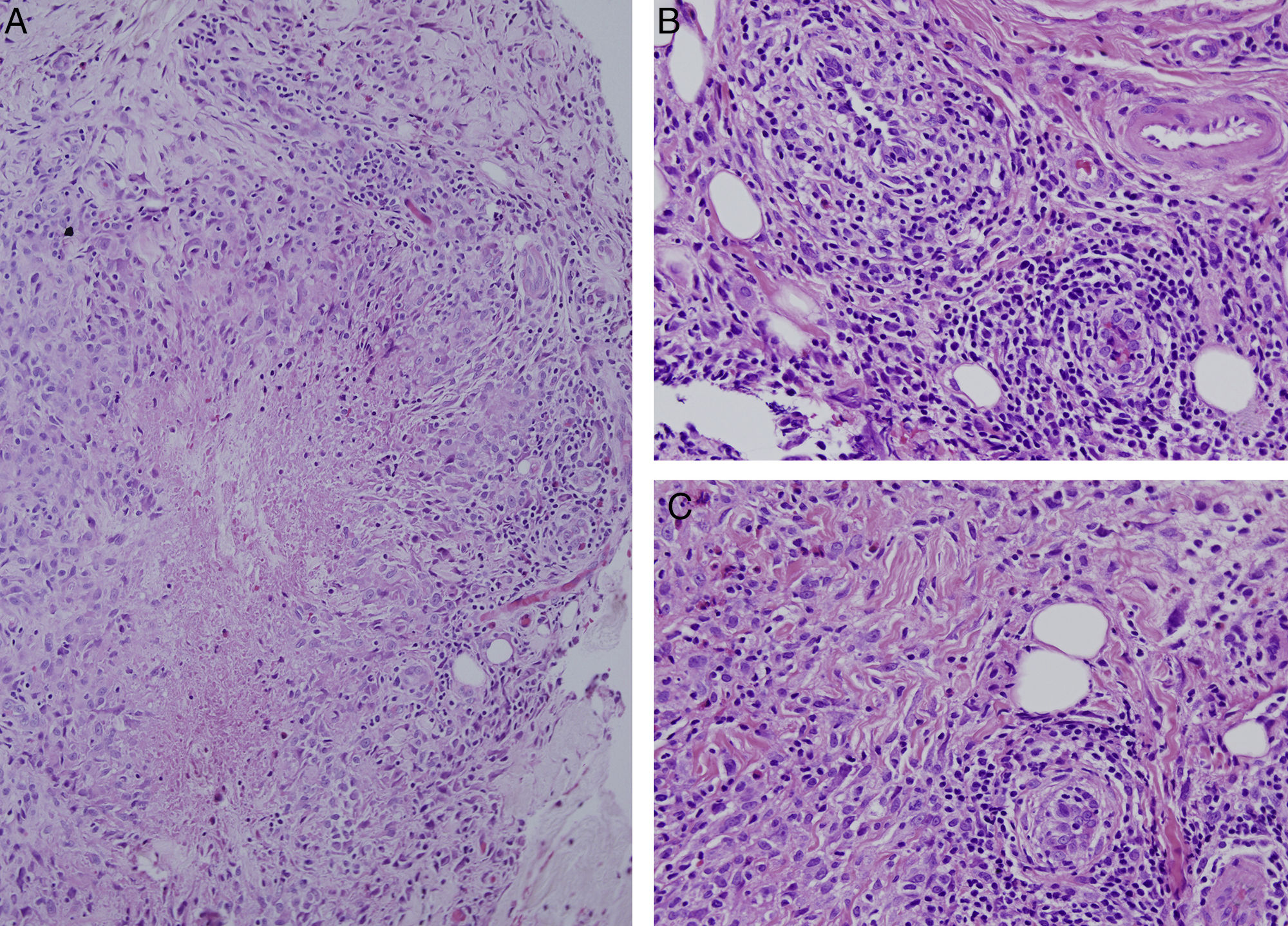
Ante la inespecificidad de los resultados de las pruebas de imagen, se realizó biopsia excisional de la lesión. El estudio histopatológico evidenció la presencia de granulomas de centro necrótico de forma estrellada o geográfica, constituidos por células epitelioides dispuestas en empalizada y entremezcladas con linfocitos (fig. 2A), así como un infiltrado inflamatorio constituido fundamentalmente por linfocitos dispuestos en torno a vasos de pequeño y mediano calibre, en ocasiones penetrando su pared y alcanzando las células endoteliales, sin presencia de necrosis fibrinoide (fig. 2B y C). Se observan también algunas células gigantes multinucleadas de citoplasma estrellado, y un infiltrado de neutrófilos (fig. 2A).
. Se asocia además un infiltrado inflamatorio constituido fundamentalmente por linfocitos dispuestos en torno a vasos de pequeño y mediano calibre en ocasiones penetrando su pared y alcanzando las células endoteliales, sin presencia de necrosis fibrinoide (B y C), que son sugestivos de lesiones de granulomatosis con poliangeitis (granulomatosis de Wegener).")
Estudio histopatológico. Se muestra la presencia de granulomas de centro necrótico de forma estrellada o geográfica, constituidos por células epitelioides dispuestas en empalizada y entremezcladas con linfocitos, así como un infiltrado de neutrófilos (A). Se asocia además un infiltrado inflamatorio constituido fundamentalmente por linfocitos dispuestos en torno a vasos de pequeño y mediano calibre en ocasiones penetrando su pared y alcanzando las células endoteliales, sin presencia de necrosis fibrinoide (B y C), que son sugestivos de lesiones de granulomatosis con poliangeitis (granulomatosis de Wegener).
El diagnóstico anatomopatológico fue de granulomatosis con poliangeitis (granulomatosis de Wegener), recomendándose descartar la afectación de otros órganos.
En la TAC pulmonar se observó un sutil patrón en vidrio deslustrado subsegmentario de dudoso significado patológico. El estudio inmunológico (ANCA, anticuerpos anti-mieloperoxidasa y anticuerpos anti-proteinasa 3) resultó negativo.
A tenor de los resultados se inició tratamiento con dosis altas de prednisona asociado a una pauta ascendente de metotrexato hasta 20mg semanales. Durante los 4 meses siguientes, la paciente fue evaluada mensualmente, procediéndose, ante una respuesta parcial, al incremento progresivo de la dosis de metotrexato, así como un descenso paulatino de la dosis de prednisona. Posteriormente se inició tratamiento con rituximab con buena respuesta y ausencia de enfermedad tanto clínica como radiológica tras 2 años de tratamiento.
Considerando la afectación orbitaria como posible manifestación precoz de la GPA, y debido a la variabilidad de las opciones diagnósticas posibles, se hace imprescindible para el diagnóstico de la enfermedad una correlación clínico-histopatológica. En estos casos la biopsia resulta imprescindible para el diagnóstico, ya que la pobre sensibilidad de otras herramientas diagnósticas de manera aislada en las formas incipientes de esta enfermedad, puede suponer un retraso en el diagnóstico y en el abordaje terapéutico.
