







La prevalencia de las displasias óseas se calcula en un caso por cada 1.000 habitantes, lo que evidencia que, en algún momento de la trayectoria profesional de un especialista en cirugía ortopédica, se encontrará ante un paciente afecto por ellas. El objetivo de este trabajo es revisar los aspectos generales de las displasias óseas y centrarnos en las que, por su frecuencia e importancia, hemos considerado más destacadas (acondroplasia, displasia epifisaria múltiple, displasia espondiloepifisaria y osteogénesis imperfecta), revisando sus características fundamentales y los últimos avances terapéuticos. No existe tratamiento curativo para estas enfermedades, por lo que el diagnóstico precoz y el manejo terapéutico adecuado se convierten en la clave para mejorar la calidad de vida de estos pacientes.
The prevalence of bone dysplasias is estimated to be one case per 1,000 inhabitants, which suggests that, at some point in the career of an orthopaedic surgeon, he will face with one of these patients. The aim of this paper is to review the general aspects of bone dysplasias and focus on those, which due to their frequency and importance, we consider most relevant (achondroplasia, multiple epiphyseal dysplasia, spondyloepiphyseal dysplasia, osteogenesis imperfecta), reviewing their fundamental features and the latest therapeutic advances. There is no cure for these diseases, so early diagnosis and appropriate therapeutic management, becomes the key to improving quality of life of these patients.
Los términos displasias óseas, displasias esqueléticas y osteocondrodisplasias se refieren a un grupo heterogéneo de trastornos del esqueleto óseo que poseen la característica común de presentar una alteración generalizada del tejido óseo y cartilaginoso. Son una de las causas más frecuentes de retraso severo del crecimiento.
Es necesario distinguir una serie de conceptos, que tienden a usarse indistintamente:
- •
Displasia: Procesos que implican defectos generalizados del esqueleto provocados por alteraciones intrínsecas.
- •
Disóstosis: Procesos limitados a un segmento óseo o hueso concreto.
- •
Distrofia: Defectos causados por un proceso extrínseco.
Existen más de 200 displasias óseas, algunas de ellas muy poco prevalentes. La frecuencia general de estas enfermedades es difícil de establecer, pero si se incluyen las formas larvadas que no llegan a diagnosticarse, su prevalencia alcanzaría un caso por cada 1.000 habitantes, siendo la acondroplasia es la más frecuente de todas ellas (fig. 1).

La patogénesis de estos trastornos puede atribuirse a un déficit de componente tisular normal, a un exceso de elementos mutantes, a la acumulación de metabolitos intermediarios o a irregularidades en los mecanismos que regulan el desarrollo del tejido osteocartilaginoso1. La etiopatogenia de estas enfermedades suele estar relacionada con mutaciones en los genes que codifican proteínas colágenas. Se han encontrado mutaciones en el gen COL2A1 en las condrodisplasias con afectación ocular como la displasia congénita espondiloepifisaria o el síndrome de Kniest, mutaciones del gen COL10A1 en la displasia metafisaria de Schmid y del COL9A2 en la displasia epifisaria múltiple (DEM). Otra proteína afectada frecuentemente en estas entidades es el receptor 3 del factor de crecimiento fibroblástico (FGFR3), que aparece mutado en la acondroplasia, la hipocondroplasia y la displasia tanatrópica. La mayoría de las displasias óseas siguen un patrón de herencia autosómica dominante. Sin embargo, muchas de ellas surgen por mutaciones de novo, siendo la edad paterna avanzada un factor de riesgo para ello.
La clasificación clásica es la de Rubin2, que las distingue en hipo o hiperplasias en función de la zona del hueso afecta (fig. 2). No obstante, hoy en día se prefiere agruparlas según el origen molecular de las mismas y los desórdenes genéticos hallados3, un ejemplo es la clasificación de Nelson4:
- •
Afectación de las proteínas de la matriz del cartílago: Displasias espondiloepifisarias, DEM.
- •
Afectación de los receptores transmembrana: Grupo de la acondroplasia.
- •
Alteración del transporte de iones: Displasia diastrófica, acondrogénesis.
- •
Afectación de los factores de transcripción: Displasia campomélica, síndrome uña-rótula.
- •
Defectos en la reabsorción ósea: Osteopetrosis.
- •
Defectos desconocidos: Distrofia torácica asfixiante o síndrome de Jeune, enfermedad de Caffey o hiperostosis cortical infantil.

En la práctica clínica diaria es más útil clasificarlas por zona esquelética afecta: a) Esqueleto axial (displasia espondiloepifisaria [DEE] y displasia metatrópica o síndrome de Kniest), b) epífisis (DEM, displasia epifisaria hemimélica) y c) metáfisis (acondroplasia, condrodisplasia tanatofórica)2,5.
A continuación se resumen las características más importantes de las displasias óseas más prevalentes1–5 y significativas en nuestro medio.
AcondroplasiaGeneralidadesEs la displasia ósea más frecuente, con una incidencia estimada en 1/30.000 recién nacidos vivos2,5, y la forma de enanismo mejor conocida1–5. El término empleado para la enfermedad no es totalmente apropiado debido a que se trata de un fallo en la osificación endocondral, y no en la condrogénesis2.
EtiologíaAunque posee un carácter autosómico dominante, hasta en el 80% de los casos surge de novo4 por una mutación puntual en el codón 380 del gen que codifica el FGFR3 situado en el brazo corto del cromosoma 41–5. El FGFR3 parece limitar la osificación endocondral, y su mutación incrementa esta inhibición2, permaneciendo inalteradas las formas de osificación intramembranosa y periostal.
CaracterísticasGeneralmente se diagnostica al nacimiento, o prenatalmente3, por la suma de talla baja desproporcionada con displasia craneofacial característica (macrocefalia, prominencia frontal y biparietal, hipoplasia mediofacial, aplastamiento del occipucio, puente nasal deprimido y prognatismo1–5). En el periodo neonatal es necesario diferenciarla de otras formas de enanismo1. En este caso es de tipo rizomélico, donde los segmentos proximales de las extremidades son relativamente cortos en comparación con el tronco1–5. Otras características presentes al nacimiento son: Pliegues cutáneos redundantes con apariencia de excesivo tejido muscular; laxitud extrema de todas las articulaciones excepto del codo, el cual presenta cierta limitación a la extensión; manos cortas y con deformidad «en tridente», por existir una separación evidente entre los dedos medio y anular, y «en estrella de mar», por tener la misma longitud los 3 dedos centrales. Durante la lactancia suele haber un retraso en la adquisición de habilidades motoras debido a la hipotonía, a la hiperlaxitud articular y a la dificultad mecánica para equilibrar una cabeza de gran tamaño con un tronco normal y unas extremidades cortas. Sin embargo, este retraso motor también puede ser la primera manifestación de una mielopatía cervical a consecuencia de una estenosis del foramen magno, entidad que es necesaria descartar siempre en estos pacientes1,2,4. Pueden presentar una cifosis toracolumbar que se convierte, conforme el niño comienza a caminar, en hiperlordosis lumbar1–5. Puede aparecer hidrocefalia, que no suele requerir tratamiento, otitis medias de repetición, que pueden derivar en sordera, y apiñamiento dental. El coeficiente intelectual de estos pacientes suele ser normal. En la infancia y en la adolescencia aparecen deformidades angulares en ambos miembros inferiores: genu varo, torsión tibial interna y desviación en varo del tobillo, debidas a la malformación tibial y a la longitud aumentada del peroné con respecto a la tibia1–5. En la edad adulta pueden aparecen coxalgia y síntomas neurológicos en los miembros inferiores como posible manifestación de una estenosis del canal raquídeo, y que no hay que achacar a osteoartritis precoz, la cual es infrecuente1.
Los hallazgos radiológicos presentes en el nacimiento son diagnósticos junto con el fenotipo; es imprescindible solicitar estudios radiográficos de cráneo, columna vertebral lumbar y pelvis1 (fig. 3). Los huesos craneales son grandes mientras que los de la base y los huesos faciales son hipoplásicos. En la columna cervical es frecuente la occipitalización del atlas1. Los pedículos vertebrales son cortos y la distancia interpedicular está invertida1,2,4. Los ilíacos son cortos y redondeados, con aspecto de «orejas de elefante», el sacro se encuentra horizontalizado y los techos acetabulares son planos. No es infrecuente un acuñamiento de T12-L1 que acaba produciendo un síndrome de cauda equina1. Todos los huesos tubulares son cortos excepto el peroné, que es desproporcionadamente largo con respecto a la tibia. La placa de crecimiento del fémur distal presenta una forma característica en «V» invertida1. La cabeza del radio se encuentra deformada e inclinada hacia la parte posterior, lo cual degenera en subluxación y/o luxación posterior de la misma en la infancia tardía.
Tratamiento
Es muy importante el diagnóstico precoz, la valoración neurológica continua (por estenosis del foramen magno en la infancia precoz y lumbar en la edad adulta) y proporcionar a la familia información sobre la enfermedad.
El uso de hormona del crecimiento no ha aportado grandes beneficios2,3, debido a que la enfermedad no es consecuencia de un déficit de la misma2, y solo se obtienen buenos resultados en aquellos pacientes con una menor velocidad de crecimiento2. Estudios recientes muestran una disminución en la expresión celular del receptor de la hormona paratiroidea y una tendencia incrementada de la apoptosis de los condrocitos, hecho que explicaría la falta de respuesta a la terapia con hormona del crecimiento2,6.
El tratamiento ortopédico está centrado en 3 aspectos: elongación ósea, corrección de las desviaciones axiales y tratamiento de la estenosis raquídea y de la cifosis toracolumbar. La elongación ósea es la que plantea mayor controversia y, aunque ha sido practicada frecuentemente en los últimos años, en la actualidad han vuelto las dudas acerca de los beneficios reales que aporta. En la práctica clínica este proceso solo proporciona ventajas si se consigue una altura final de unos 150cm, lo cual implica elongaciones de 25-30cm en miembros inferiores, y la necesidad de alargar también los húmeros para facilitar el cuidado personal. El tiempo total requerido puede exceder los 2 años, y el riesgo de complicaciones es elevado. Diversos autores no han encontrado diferencias estadísticamente significativas en cuanto a la mejora de la calidad de vida en pacientes elongados en comparación con pacientes acondroplásicos que no se han sometido a esta cirugía7–9. Seung-Ju et al.10 emplearon 3 cuestionarios, constatando que las puntuaciones obtenidas en los componentes mental, físico y funcional eran similares para ambos grupos de pacientes y que dichas puntuaciones eran inversamente proporcionales al número de complicaciones surgidas durante el proceso de elongación. Estos autores concluyeron que la elongación ósea podía ser una opción razonable de tratamiento siempre que los pacientes comprendan los beneficios y riesgos del proceso y de que se trate de un proceso multidisciplinar que preste especial atención a las complicaciones que vayan surgiendo para actuar precozmente sobre ellas. La asociación Little People of America llegó a estas mismas conclusiones11.
En cuanto al tratamiento de las deformidades raquídeas, es la cifosis la más frecuente, fundamentalmente a nivel D12-L1, y la estenosis la más grave1–5. En la mayoría la cifosis mejora cuando el niño comienza a caminar, pero existe un 10-15% de los casos en los cuales permanece e incluso se incrementa. El tratamiento dependerá de la edad en que se detecte la deformidad. Durante la infancia el objetivo es prevenir la aparición de la deformidad mediante determinadas normas posturales tales como evitar que los afectados se sienten en sillas sin respaldo y lograr que mantengan la espalda recta en su vida cotidiana. Una vez establecida, el corsé está indicado si la cifosis se acompaña de un acuñamiento vertebral progresivo y significativo y/o si no se reduce el ángulo de Cobb por debajo de 30° en radiografías en hiperextensión. En los casos en los que el corsé sea insuficiente, puede aplicarse un corsé de yeso en hiperextensión. Los casos en los que la cifosis persista a pesar del tratamiento conservador, tendrán indicación de tratamiento quirúrgico. El diagnóstico de estenosis raquídea, normalmente realizado durante la tercera década de la vida por la presencia de claudicación neurógena, es una indicación absoluta e inmediata de descompresión quirúrgica. Esta se efectúa desde varios niveles por encima de la zona estenosada hasta S2, siendo necesario realizar laminectomías e incluso foraminotomías en aquellos casos en los que estén comprometidas también las raíces2. Una vez establecida esta complicación, los resultados son impredecibles1.
Actualmente se encuentran abiertas varias líneas de investigación que intentan reducir la hiperactividad del FGFR312, destacando: la inhibición química selectiva de la actividad tirosina-cinasa del FGFR313, anticuerpos que bloquean la activación del FGFR3 basados en el existente trastuzumab14,15 (anticuerpo monoclonal anti HER2/neu) y el uso del péptido natriurético tipo C como antagonista de la señal desatada por FGFR316–18.
A pesar de que esta enfermedad se asocia a una alta tasa de mortalidad perinatal, los pacientes que sobreviven poseen una esperanza de vida similar a la de la población general4 con un índice de valoración global de su salud no significativamente disminuido2. La talla media final alcanzada es de 131cm en hombres y de 124cm en mujeres1.
Displasia epifisaria múltipleGeneralidadesLa DEM es otra de las displasias óseas más frecuentes, producida por una disrupción en la osificación endocondral de las epífisis, sobre todo a nivel de la epífisis femoral proximal3. Presenta una prevalencia de unos 10-12 casos por millón de habitantes4. Clásicamente, se distinguían 2 variantes de la enfermedad, la forma severa o tipo Fairbank, y una variante más leve, la de Ribbing. Actualmente, y en función de los conocimientos actuales de las bases genéticas de esta displasia ósea, se acepta una única enfermedad sin variantes, pero con una expresión muy variable2.
EtiologíaEs un desorden autosómico dominante, aunque existen descritos en la literatura casos recesivos de la enfermedad, causados por una mutación en el gen que codifica la proteína oligomérica de la matriz del cartílago situada en el cromosoma 19. También existen mutaciones en los genes que codifican el colágeno tipo ix (COL9A2) y la matrilina-31,2,4. El resultado final de estas mutaciones es una anomalía en la matriz de los cartílagos hialino, articular y fisario1 que resulta en las deformidades típicas de la enfermedad.
CaracterísticasNo suele ser hasta la infancia precoz cuando se diagnostica la DEM por una de las siguientes consultas: dolor articular en las extremidades inferiores (fundamentalmente las caderas), disminución del rango de movilidad, alteraciones de la marcha, o deformidades angulares de las rodillas2. Las articulaciones de carga (caderas, rodillas y tobillos) son las que se afectan con mayor frecuencia. Las manos de los pacientes son pequeñas y con alteraciones de la movilidad, siendo la fatiga de las manos al escribir un rasgo típico1. La velocidad de crecimiento se encuentra disminuida, con resultado final de talla corta, aunque sin llegar al enanismo1–3. La facies y la columna vertebral son normales. Tampoco hay alteraciones viscerales ni afectación del nivel de inteligencia. En los casos familiares relacionados con la proteína oligomérica de la matriz del cartílago es posible el diagnóstico prenatal.
La radiología convencional es fundamental para el diagnóstico de la enfermedad y son necesarios estudios de pelvis, columna vertebral, rodillas, tobillos y muñecas1. Como hallazgos radiológicos puede haber retraso en la aparición de cualquier núcleo de osificación, aunque los cambios serán más evidentes en las epífisis proximales y distales de los fémures, y proximales de los húmeros y tibias2. La severidad, así como la extensión de la enfermedad, son muy variables entre familias, lo cual influye fundamentalmente en la edad de aparición de la osteoartrosis. Los cambios radiológicos en las caderas recuerdan mucho a los que aparecen en la enfermedad de Perthes, principal diagnóstico diferencial de esta displasia ósea. De hecho, en cualquier paciente sospechoso de padecer un Perthes bilateral, debe descartarse una DEM1–3. Los rasgos que diferencian esta entidad de la enfermedad de Perthes son: Los cambios radiológicos en caderas son simétricos y bastante sincrónicos, respeto de las metáfisis femorales y del cartílago articular, afectación más severa del acetábulo y ausencia de subluxación de las cabezas femorales2. La DEE es la otra displasia ósea de la que hay que diferenciar la DEM, siendo la afectación o no de la columna vertebral el punto clave para distinguirlas1. La osteoartrosis es el principal problema de estos pacientes y suele presentarse entre la segunda y tercera décadas de la vida, dependiendo, como hemos comentado antes, de la severidad de la enfermedad.
TratamientoEl tratamiento es sintomático, y los aspectos claves del mismo son el diagnóstico precoz y el seguimiento1–5. El reposo y la descarga con bastones están indicados en la coxalgia durante la infancia. El uso de paracetamol está recomendado en aquellos pacientes con aumento nocturno del dolor de cadera1. No está demostrado que las osteotomías femorales mejoren el pronóstico ni la necesidad futura de artroplastia total de cadera3 (fig. 4). De hecho, en pacientes con un grado importante de coxa vara preexistente, se encuentran incluso contraindicadas2. La artroplastia total de cadera bilateral a edades tempranas será la solución definitiva para la mayoría. En algunos, puede ser necesaria también la artroplastia glenohumeral.

La esperanza de vida es normal y su limitación fundamental es la coxartrosis precoz. La talla final oscila entre 145 y 170cm de altura.
Displasia espondiloepifisariaCaracterísticasLa DEE incluye un conjunto de trastornos cuya característica principal es el retraso en la osificación de las epífisis de huesos largos así como del esqueleto axial. La prevalencia está estimada en 7-11 casos por millón de habitantes. Distinguimos varios trastornos dentro de este grupo como la acondrogénesis tipo ii y la hipocondrogénesis, letales ambas en el recién nacido, y las formas congénita y tardía de la DEE propiamente dicha.
Displasia espondiloepifisaria congénitaCon una prevalencia de 3-4 casos por cada millón de habitantes2, es un trastorno autosómico dominante aunque la mayoría de los nuevos casos surgen por mutaciones de novo2,3 en el gen que codifica el colágeno tipo ii, situado en el brazo largo del cromosoma 12, y que genera una alteración de la función de esta proteína en los cartílagos de crecimiento y articular, en los discos intervertebrales y en el humor vítreo1,3. Se trata de una patología evidente al nacimiento por el fenotipo característico: enanismo rizomélico, aplanamiento de la cara con hipoplasia malar y paladar hendido, pectus carinatum y pie zambo1–5. Durante la infancia, es frecuente la hipotonía y el retraso del desarrollo motor, y es obligatorio descartar que la causa subyacente sea una mielopatía cervical causada por una inestabilidad atlantoaxoidea, frecuente en estos pacientes. Otra complicación destacable en este periodo es la insuficiencia respiratoria a consecuencia del pequeño tamaño de la cavidad torácica2,4. La lordosis lumbar, el desarrollo de una cifoescoliosis progresiva y la marcha de pato por coxa vara y contractura en flexión de las caderas son rasgos comunes durante los primeros años de vida del paciente. Durante la adolescencia y la edad adulta comienza a aparecer una osteoartrosis precoz sobre todo a nivel de caderas y rodillas. Como alteraciones extraesqueléticas, existe un elevado riesgo de desprendimiento de retina durante la adolescencia y la posibilidad de desarrollar problemas de audición1,2,4. Las anomalías radiológicas se encuentran presentes al nacimiento y consisten en un retraso en prácticamente todos los centros de osificación1–5, siendo los más afectados el fémur y la tibia proximales, la pelvis y la columna vertebral1. A nivel raquídeo, son comunes la hipoplasia de la odontoides o la presencia de un os odontoideum, el estrechamiento del espacio intervertebral, la hiperlordosis lumbar progresiva y la cifoescoliosis torácica con una angulación brusca concentrada en varios cuerpos vertebrales2. Al no producirse la osificación de la epífisis femoral proximal durante la infancia, aparece coxa vara progresiva, ascenso del trocánter mayor y acortamiento del cuello femoral1,2, todo lo cual, unido al flexo de caderas, contribuye a la osteoartrosis precoz. En cuanto al manejo de la enfermedad, lo primero es realizar un diagnóstico diferencial con algunas formas de mucopolisacaridosis, enfermedad de Morquio fundamentalmente, con el hipotiroidismo y con la seudoacondroplasia3. Para establecer el diagnóstico, así como para el posterior seguimiento, es prioritaria la realización de un mapa óseo completo. La complicación más grave es la mielopatía cervical por inestabilidad atlantoaxoidea, cuya sospecha obliga a la realización de una exhaustiva exploración neurológica, radiografías cervicales en flexión y extensión e incluso una resonancia magnética1,2. Algunas de las opciones terapéuticas son la fusión atlantoaxoidea, la descompresión quirúrgica o la fusión del atlas al occipucio2. Respecto a la escoliosis, presente en más de la mitad de los pacientes, si la curva es menor de 40° se debe intentar controlar mediante un corsé2, quedando reservada la corrección quirúrgica para curvas mayores. La cifosis puede controlarse de manera óptima mediante un corsé tipo Milwaukee siempre que este se mantenga hasta la madurez ósea2. En prácticamente la totalidad de los pacientes será preciso recurrir a osteotomías tanto femorales como acetabulares para el tratamiento de la coxa vara, recomendándose asimismo el tratamiento conjunto de las contracturas en flexión2. La osteotomía intertrocantérica en Y de Pauwel está especialmente indicada en estos pacientes1. Debe evaluarse la alineación de las rodillas antes de proceder a la corrección de las caderas, y corregirse en el mismo acto quirúrgico si fuera necesario. En la mayoría de los pacientes, la sustitución protésica de las articulaciones afectas será irremediable a una edad temprana1–5 precisando, en muchos casos, nuevas osteotomías concomitantes. Las deformidades de los pies se tratan de la misma forma que en pacientes no afectos por esta enfermedad2. La talla final de los pacientes está comprendida entre 80 y 125 cm1,2.
Displasia espondiloepifisaria tardíaEs un trastorno ligado al cromosoma X, por tanto más grave en varones, aunque se ha descrito una forma recesiva. Está causada por una mutación en el colágeno tipo ii2. Las manifestaciones clínicas no aparecen hasta la infancia, cuando se hace evidente la talla corta acompañada de dolor en caderas, rodillas y espalda. Los rasgos típicos de esta enfermedad son el tronco acortado, el pectus carinatum y la osteoartrosis prematura, particularmente en caderas y hombros1–5. La talla final oscila en torno a los 150cm. Radiológicamente se aprecia afectación predominantemente de los hombros, caderas y rodillas, y platispondilia generalizada. Puede existir hipoplasia de odontoides o también os odontoideum, causas de inestabilidad atlantoaxoidea, la cual hay que descartar2, y de escoliosis. Es necesaria la asociación clínica y radiológica para establecer el diagnóstico de la enfermedad y realizar el diagnóstico diferencial con la DEM (no suele afectar al esqueleto axial) y con la enfermedad de Scheuermann (la afectación vertebral es generalizada)1. La sustitución protésica total, precedida o no de osteotomías correctoras, será necesaria en la mayoría de los pacientes a edades más tempranas que en la población general1–5.
Osteogénesis imperfectaLa osteogénesis imperfecta (OI) es la causa más frecuente de osteoporosis de origen genético4. Posee una incidencia calculada de 1:20.000 recién nacidos vivos1–5, aunque existe una mutación descrita en África Occidental del tipo viii de la enfermedad con una incidencia de portadores de 1:200-300 afroamericanos4.
Posee una enorme variabilidad, cuyo origen son defectos cuantitativos y/o estructurales en el colágeno tipo i, componente principal de la matriz extracelular del hueso y de la piel4. La mayoría de los casos siguen un patrón de herencia autosómica dominante, ya sea por mutaciones preexistentes o de novo, aunque esta entidad también puede heredarse de manera autosómica recesiva1,3,4. Las formas autosómicas dominantes pueden estar causadas por 2 tipos de mutaciones sobre los genes que codifican y ensamblan el colágeno tipo i: inactivación de uno de los alelos del gen, resultando en una reducción del 50% de la cantidad de colágeno, estructuralmente normal, o reducción de la cantidad de colágeno tipo i asociada a la mutación de alguna de sus cadenas, existiendo una reducción cuantitativa y estructural del colágeno1. El resultado es la síntesis de un hueso con trabéculas finas y fibrosas y con un contenido anormal de proteoglicanos y de la sustancia fundamental3 muy sensible a la rotura y a las deformidades.
Clínicamente, la enfermedad se caracteriza por la tríada de Eddowes van der Hoeve: Fragilidad ósea, escleróticas azules y sordera temprana4,19. Se diferencian actualmente 8 tipos de esta enfermedad, basados en los 4 tipos descritos por Sillence en 19791,4,20,21 según criterios clínicos y radiológicos:
Tipo I. La forma clásica de la OI, es la más común y la más leve1. Se transmite de forma autosómica dominante y los pacientes presentan la tríada clásica. Se diferencian 2 subtipos, A y B, según presenten (B) o no (A) dentinogénesis imperfecta1,4. Las fracturas son más frecuentes que en niños sanos y aparecen cuando el niño comienza a caminar, disminuyendo su frecuencia tras la pubertad. Posteriormente, la incidencia de fracturas vuelve a aumentar en la edad adulta1,4. La consolidación ósea de las fracturas sigue un proceso normal.
Tipo II. Con 2 subtipos, es la forma más severa de la enfermedad, letal prenatalmente o durante el primer año de vida1,3,4.
Tipo III. Se trata de la forma más grave no mortal y la de mayor interés ortopédico19. Los recién nacidos presentan múltiples fracturas intraútero y al nacimiento, con un tórax ensanchado, al contrario de lo que ocurre en la tipo ii. Las fracturas consolidan de forma viciosa, contribuyendo a aumentar las deformidades esqueléticas existentes1,4. Se distinguen 2 subtipos: de herencia autosómica dominante con mutaciones en genes codificadores del colágeno tipo i, y recesiva con mutaciones en genes relacionados con CRTAP1. Aunque el peso y la longitud al nacer son casi normales, la estatura alcanzada por estos pacientes es extremadamente baja. Es frecuente la asociación con dentinogénesis imperfecta. Los huesos largos tienden a arquearse progresivamente, las epífisis son grandes en comparación con las diáfisis y en las metáfisis aparecen islas de osificación endocondral que le aportan un aspecto en «palomitas de maíz» en la radiología convencional1,4. Casi todos presentan además cifoescoliosis y compresión vertebral por el aplastamiento progresivo de los cuerpos vertebrales. Los niños pueden conseguir caminar durante un periodo de tiempo, aunque la mayoría dejará de hacerlo con el crecimiento1.
Tipo IV. Es similar al i, aunque algo más grave y con escleróticas normales1,4. La mayoría no sufren grandes deformidades. Con la deambulación, en general conseguida por todos, comienzan a producirse nuevas fracturas, cuya frecuencia disminuye tras la pubertad. Radiológicamente existe osteoporosis, ensanchamiento metafisario y compresión vertebral. El principal diagnóstico diferencial de esta forma es con el niño maltratado1,3–5.
Tipo V. Clínicamente similar al anterior pero con hallazgos radiológicos e histológicos distintos4. Autosómica dominante, su defecto genético es aún desconocido1. Las radiografías de antebrazos y piernas muestran una osificación de las membranas interóseas y bandas metafisarias radiodensas. Presentan callos de fractura hiperplásicos que incluso pueden simular un osteosarcoma1,4.
Tipo VI. Clínicamente similar al tipo iv pero más severo. No presenta mutaciones demostrables en el colágeno tipo i1. La forma de transmisión y el defecto genético son desconocidos.
Tipo VII. Se transmite de forma autosómica recesiva y suelen presentar mutaciones nulas en el P3H1 y en el CRTAP4. Al nacimiento y durante la infancia los afectados presentan múltiples fracturas cuya frecuencia disminuye en la adolescencia1. Otras características de este tipo de OI son el enanismo rizomélico, coxa vara severa y deformaciones progresivas esqueléticas.
Tipo VIII. La transmisión y la mutación son iguales que en el tipo vii y podemos describirla como un solapamiento entre las formas II y III1,4. Muchos de los pacientes fallecen en la infancia.
DiagnósticoEn la mayoría de los casos se realiza mediante clínica, historia familiar y radiología convencional1,3–5. Son las formas esporádicas del tipo iv las que resultan más difíciles de diagnosticar y frecuentemente se confunden con el síndrome del niño maltratado, precisando una evaluación cuidadosa de las radiografías o una densitometría ósea si persisten dudas para descartarla. Los estudios bioquímicos del colágeno confirman el diagnóstico aunque no se realizan rutinariamente1, y la secuenciación de ADN es útil para distinguir entre los diferentes tipos y facilita el diagnóstico prenatal y los estudios familiares4. Otros diagnósticos diferenciales a tener en cuenta son3: displasia camptomélica, donde existe arqueamiento de huesos largos, insuficiencia respiratoria y numerosas deformidades extraesqueléticas; picnodisóstosis, con una mayor incidencia de fracturas pero con densidad ósea aumentada; osteoporosis idiopática juvenil, en la cual las fracturas recidivantes suelen ser metafisarias en lugar de diafisarias, y el nuevo hueso formado es ya osteoporótico en vez de normal, denominándose el proceso de consolidación neo-osteoporosis5; y osteomalacia, la cual posee unos hallazgos bioquímicos determinados.
TratamientoNo existe tratamiento curativo de la enfermedad. La terapia génica es difícil de aplicar debido a que cada paciente posee su propia mutación responsable de su enfermedad1. El uso de hormona del crecimiento no ha aportado beneficios más que histológicos4,19. Como tratamiento médico de base se están utilizando actualmente los bifosfonatos1,2,4,5,19,21. Su uso no está exento de controversias y no se conocen con exactitud sus indicaciones precisas, ni está establecida la duración del tratamiento. Está demostrado, sobre todo en el caso del pamidronato21–26, que reducen el número de fracturas y que aumentan el volumen óseo al disminuir la resorción osteoclástica. En las publicaciones más recientes no parece existir duda alguna de la eficacia de los bifosfonatos a la hora de reducir el número de fracturas, aumentar la masa ósea y disminuir el dolor de origen esquelético en pacientes afectos de OI21,27–30, por lo que su indicación parece estar aceptada. Las preguntas que están surgiendo actualmente son cuál es la duración adecuada del tratamiento y cuáles los efectos secundarios derivados del mismo. Nicolaou et al.31–35 realizaron un estudio retrospectivo comparando las fracturas femorales ocurridas en 176 pacientes afectos de OI tratados con bifosfonatos durante 2 años, y observaron una disminución en el número de fracturas femorales en el grupo tratado con bifosfonatos, junto con un aumento de fracturas en la región subtrocantérica del fémur, muchas de ellas ocurridas con traumatismos de baja energía, como en los pacientes osteoporóticos tratados con bifosfonatos. Por ello, recomiendan evitar realizar osteotomías correctoras a este nivel y realizar screening radiográfico, sobre todo en aquellos que presenten dolor en dicha zona. Según varios estudios, la asociación de bifosfonatos con alfacacidol es más efectiva en el incremento de la densidad mineral ósea (DMO) y en la prevención de fracturas que el uso aislado de bifosfonatos36,37. Iwamoto et al. presentaron un caso de un paciente adulto con osteogénesis imperfecta tratado durante 11 años con esta combinación38. Con esta pauta se ha observado un aumento de la DMO, una ausencia de fracturas en huesos largos, y una mejoría significativa del dolor de espalda.
Por otro lado, la mayoría de los estudios realizados se han basado en el uso exclusivo del pamidronato22–26. Ingmar et al. analizaron el ibandronato en 27 pacientes afectos de OI39, concluyendo que su uso normaliza los marcadores de recambio óseo y produce un aumento en la DMO.
Las nuevas líneas de tratamiento abiertas en esta enfermedad son el trasplante de médula ósea y de células del estroma mesenquimal, y el uso del denosumab. Este último es un anticuerpo monoclonal frente a RANKL, inhibiendo la formación de osteoclastos y la degradación ósea por lo que está indicado para el tratamiento de la osteoporosis40–44. Entre los pocos estudios que analizan el uso del denosumab en OI está el de Semler et al.44 que concluyen que el uso del anticuerpo monoclonal anti-RANKL produce una supresión de la resorción ósea basada en la normalización de los marcadores bioquímicos de recambio óseo, una disminución de los niveles de calcio sérico sin sintomatología de hipocalcemia, y una disminución de la tasa de fracturas; que la degradación del fármaco se produce 3-4 meses después de su administración, al contrario de lo que ocurre con los bifosfonatos que permanecen almacenados en el esqueleto durante años; y recomiendan una pauta de administración de 2 meses para niños. Por todo ello, los autores animan a realizar nuevos estudios con este anticuerpo monoclonal en pacientes afectos de OI y dejan abierta una nueva y esperanzadora opción de tratamiento.
En cuanto al tratamiento de las fracturas, hemos de saber que el proceso de consolidación es totalmente normal1,3–5, excepto en la tipo v en la que existen callos hipertróficos. Por ello, las únicas diferencias en el manejo de las fracturas son1,4: reducir al mínimo la inmovilización para disminuir la osteoporosis producida por esta, realizar diagnósticos clínicos de las fracturas (reservando la radiología para casos dudosos) y ser lo más conservador posible en el quirófano porque los pacientes tendrán que pasar por él en más de una ocasión.
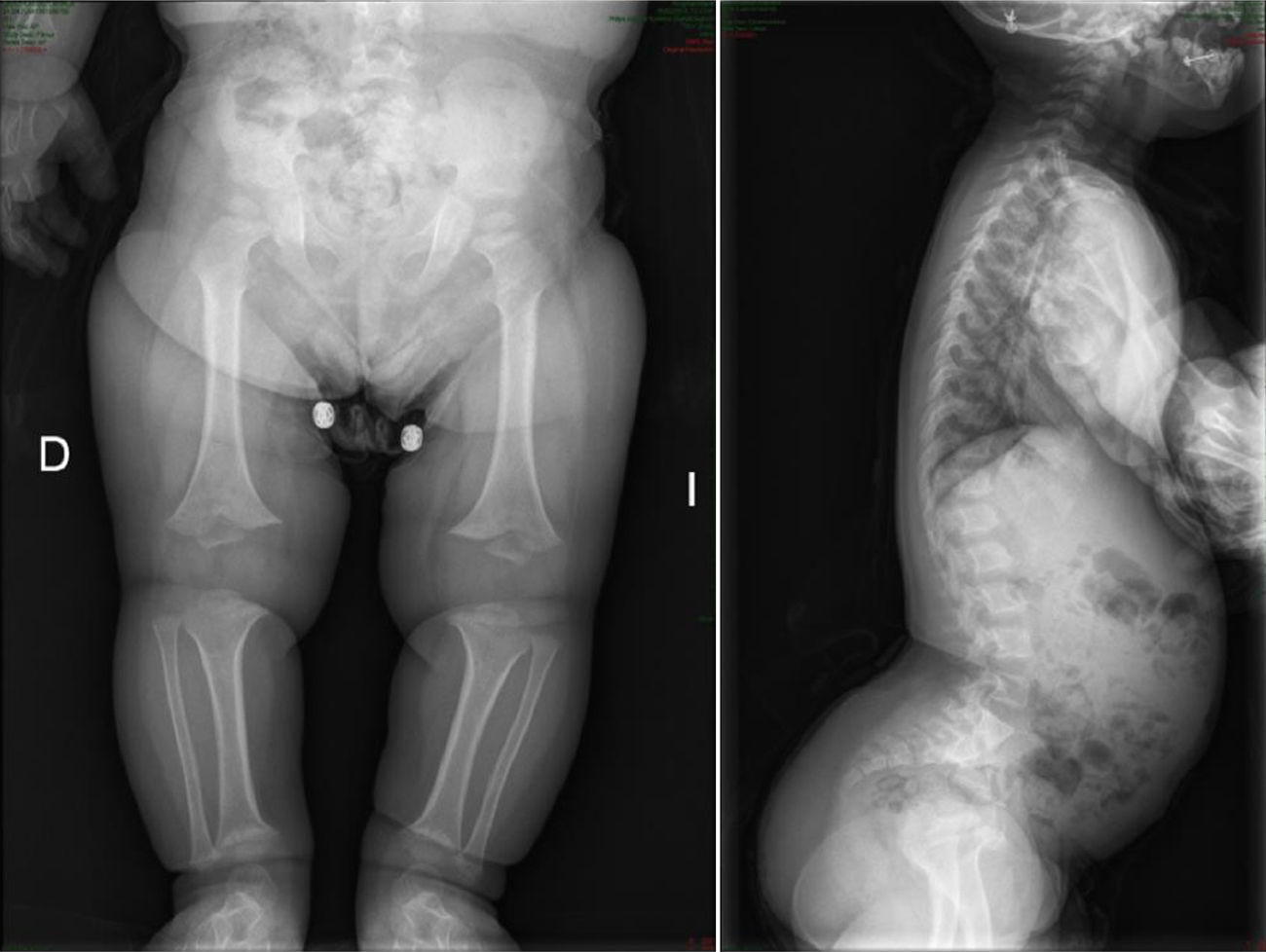
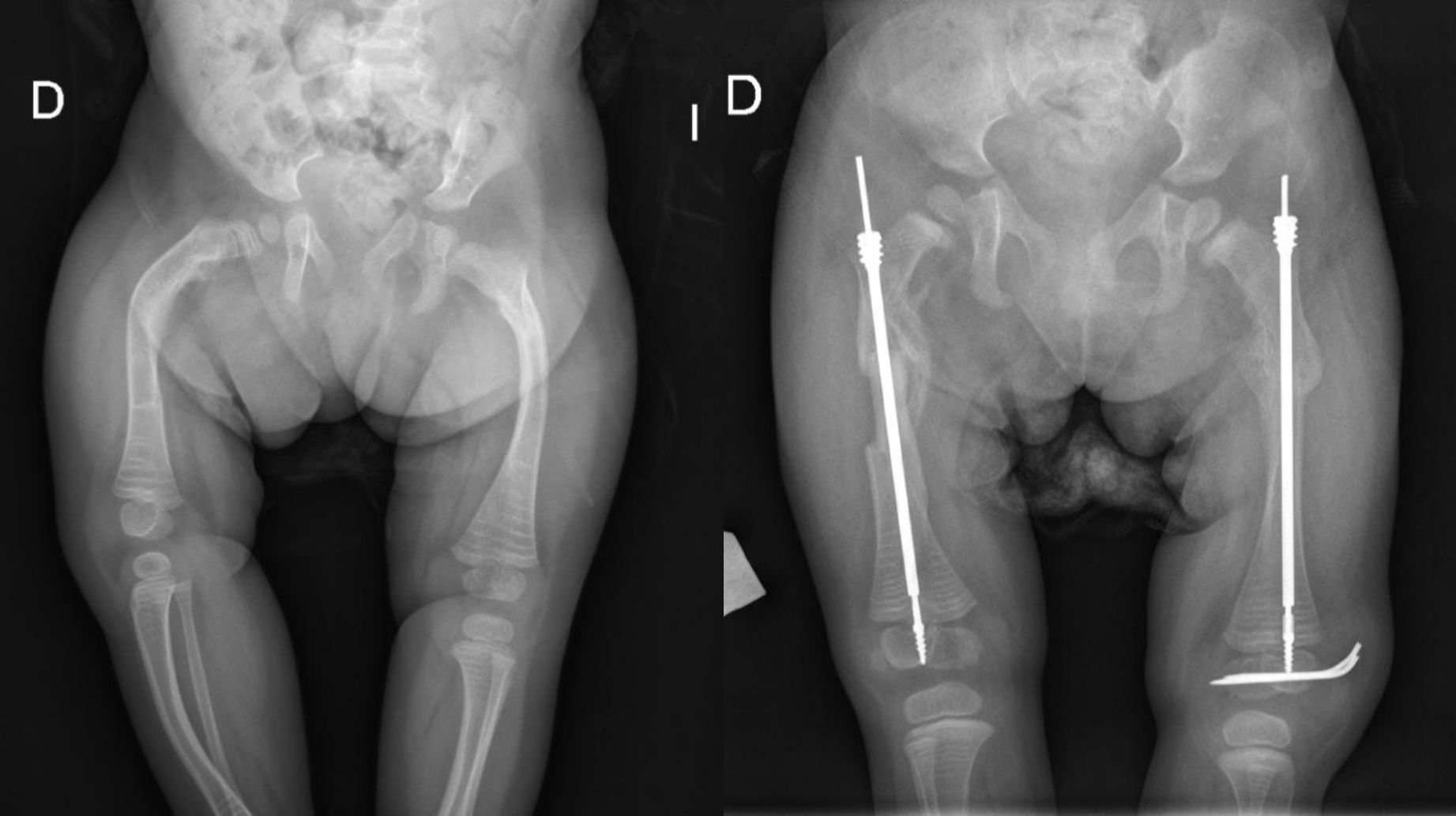
El enclavado endomedular es el tratamiento de elección para las fracturas recidivantes o las deformidades de huesos largos1,3–5. Actualmente se utilizan sistemas endomedulares que «crecen» con el hueso (figs. 5 y 6) lo que les permite estar colocados durante largos periodos sirviendo de «tutor» para evitar la aparición de nuevas fracturas.


Las displasias óseas constituyen una causa importante de retraso severo del crecimiento. Para la práctica clínica diaria es más útil clasificarlas en función de la zona del esqueleto afecta: espondilodisplasias, displasias metafisarias, displasias epifisarias y combinaciones de las anteriores. La acondroplasia es la más frecuente.
En la acondroplasia es importante descartar la sintomatología derivada de la estenosis del canal vertebral y del foramen magno, que si bien no constituye la complicación más frecuente sí es la de mayor gravedad. La elongación ósea sigue tratándose de una opción terapéutica controvertida. Las últimas líneas de investigación se centran en fármacos biológicos que disminuyan la actividad del FGFR3.
La DEM se considera en la actualidad una enfermedad con una expresividad variable. El principal diagnóstico diferencial es con la enfermedad de Perthes.
En la DEE se distinguen una forma congénita y otra tardía, en ambas encontramos afectación axial y epifisaria aunque en distinto grado. La inestabilidad atlantoaxoidea es una anomalía frecuente e importante en el diagnóstico y seguimiento de los pacientes, ya que puede ser causa de mielopatía cervical.
En la OI el proceso de consolidación de las fracturas es normal, por lo que su tratamiento es similar al de las fracturas ocurridas en niños sanos, evitando la inmovilización prolongada ya que aumenta la osteoporosis. Se diferencian 8 tipos de OI, siendo el iii la forma más grave no mortal y, por tanto, la de mayor interés ortopédico, y el iv la forma de mayores implicaciones jurídicas ya que puede confundirse con el síndrome del niño maltratado. Actualmente se usan los bifosfonatos como tratamiento de base de la enfermedad; las últimas líneas de investigación se centran en el uso del denosumab con el que se están obteniendo resultados esperanzadores.
Nivel de evidenciaNivel de evidencia v.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
