




Presentamos el caso clínico de una mujer de 80 años con antecedentes personales de dislipemia, ictus isquémico de etiología indeterminada, diabetes mellitus tipo 2, hipertensión arterial y miositis inflamatoria, en tratamiento diario con ezetimiba 10mg, ácido acetil salicílico 300mg, insulina glargina 12UI, prednisona 2,5mg, trazodona 50mg y de manera semanal metotrexato 15mg y Acfol® 5mg. En cuanto a su situación basal presentaba dependencia establecida como consecuencia de ictus previo hacía dos años (índice de Barthel 30/100, no realizaba deambulación, solo bipedestación con ayuda de 2 personas, dependencia para el aseo, la ducha, incontinencia urinaria) y sin deterioro cognoscitivo.
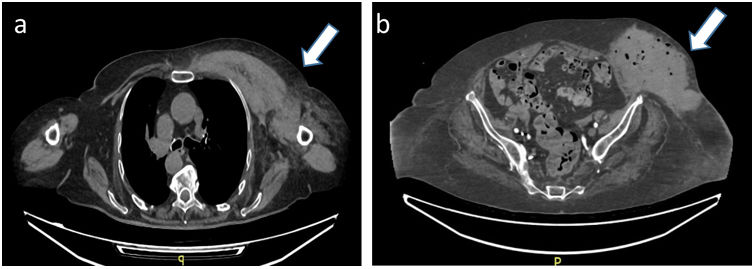
Ingresó de manera urgente en el servicio de cirugía general por hematoma espontáneo de gran tamaño en miembro superior derecho y hemitórax ipsilateral. Se realizó tomografía axial computarizada (TAC) objetivando hematoma pectoral de 9,7×5,2×5,9cm (fig. 1a) con anemia severa secundaria (hemoglobina de 6,4g/dl). Se solicitó seguimiento clínico por geriatría. Al ingreso, además de la anemia, presentaba alteración de la coagulación con tiempo de cefalina de 76s (rango normal: 20-39s) y ratio del tiempo de cefalina 2,87 (rango normal: 0,8-1,2). Una vez descartadas las causas habituales de la alteración de la coagulación, se consultó al servicio de hematología quien confirmó el diagnóstico de hemofilia A adquirida (HAA) con una determinación de FVIII del 0% mediante método cromogénico.
 Imagen de tomografía axial computarizada que muestra el hematoma espontáneo pectoral de 9,7×5,2×5,9cm; b) Imagen de tomografía axial computarizada que muestra hematoma espontáneo en pared abdominal.")
La HAA se produce por la presencia de autoanticuerpos específicos contra el factor VIII (FVIII) de la coagulación. Estos anticuerpos son policlonales, de subclase IgG4 y se unen tanto a la cadena pesada como a la ligera, impidiendo la unión del FVIII a otros factores de la coagulación1,2. En la mayoría de las ocasiones se debe a neoplasias, fármacos o enfermedades autoinmunes. Es una enfermedad de baja incidencia, con 1,5 casos/millón de habitantes/año, pero más frecuente en mayores de 85 años, entre los que la incidencia aumenta a 15 casos/millón3. La sospecha diagnóstica debe hacerse ante la aparición aguda de hematomas de tamaño o localización anormales y un alargamiento del tiempo de cefalina no justificado que no se corrige tras la realización del estudio de mezclas4. La confirmación diagnóstica se realiza con la cuantificación de niveles plasmáticos de FVIII y la titulación del inhibidor del mismo. El número y gravedad de las hemorragias no están relacionados ni con los niveles de FVIII ni con el título del inhibidor de FVIII5. No existen niveles de referencia de FVIII o de título del inhibidor para decidir cuándo iniciar el tratamiento6, sino que este depende de la localización e intensidad de la hemorragia7–9. Los objetivos del tratamiento son el control de la clínica hemorrágica mediante los llamados agentes bypass (concentrado de factores del complejo protrombínico activado y FVII activo recombinante), y la erradicación del inhibidor mediante tratamiento inmunosupresor que permita restablecer unos niveles de FVIII dentro de la normalidad. Presenta unas tasas de mortalidad elevadas, entre el 9-33%7,10.
En la paciente se cuantificó el inhibidor del FVIII mediante el test de Nijmegen-Bethesda presentado títulos elevados de 50 unidades Bethesda. Ante el diagnóstico de hemofilia adquirida con clínica hemorrágica de gravedad se inició, además de la transfusión de hemoderivados, tratamiento hemostático con FVIIa recombinante y tratamiento inmunosupresor con prednisona (1mg/kg/día), inmunoglobulinas (0,4g/kg/24h) y FVIII a dosis de 100UI/kg/día. Se descartó origen oncológico mediante TAC toraco-abdomino-pélvico y marcadores tumorales, así como origen farmacológico, por lo que se diagnosticó HAA idiopática. La paciente presentó mala evolución clínica durante los siguientes 30 días sin respuesta al tratamiento inmunosupresor, con múltiples complicaciones médicas: nuevos episodios hemorrágicos en pared abdominal (fig. 1b) que precisaron drenaje percutáneo, sobreinfección del mismo, bacteriemia secundaria por germen multirresistente, insuficiencia cardiaca descompensada, desnutrición proteica, síndrome confusional agudo e inmovilismo. Dada la evolución clínica tórpida a pesar de tratamiento dirigido y situación de dependencia previa, se decidió de manera multidisciplinar, en consenso con paciente y familiares adecuación del esfuerzo terapéutico falleciendo a las 2 semanas con buen control sintomático.
Consideramos de interés este caso porque la baja incidencia de esta enfermedad puede dificultar y retrasar el diagnóstico. A pesar de ser una enfermedad grave y con alta morbimortalidad, es una afección poco conocida donde la sospecha clínica es fundamental para el diagnóstico y tratamiento precoces.
