




El síndrome antisintetasa (SA) es una enfermedad sistémica infrecuente incluida en el grupo de las miopatías inflamatorias caracterizado por afectación pulmonar intersticial, poliartritis no erosiva, miositis, «manos de mecánico», fiebre y fenómeno de Raynaud, con anticuerpos dirigidos contra la sintetasa que une el aminoácido histidina con su tRNA correspondiente, siendo el más frecuente el anti-Jo-1. La ausencia de afectación muscular aunque descrita, es poco frecuente. Presentamos un caso con diagnóstico de síndrome antisintetasa en ausencia de miopatía inflamatoria en el paciente anciano.
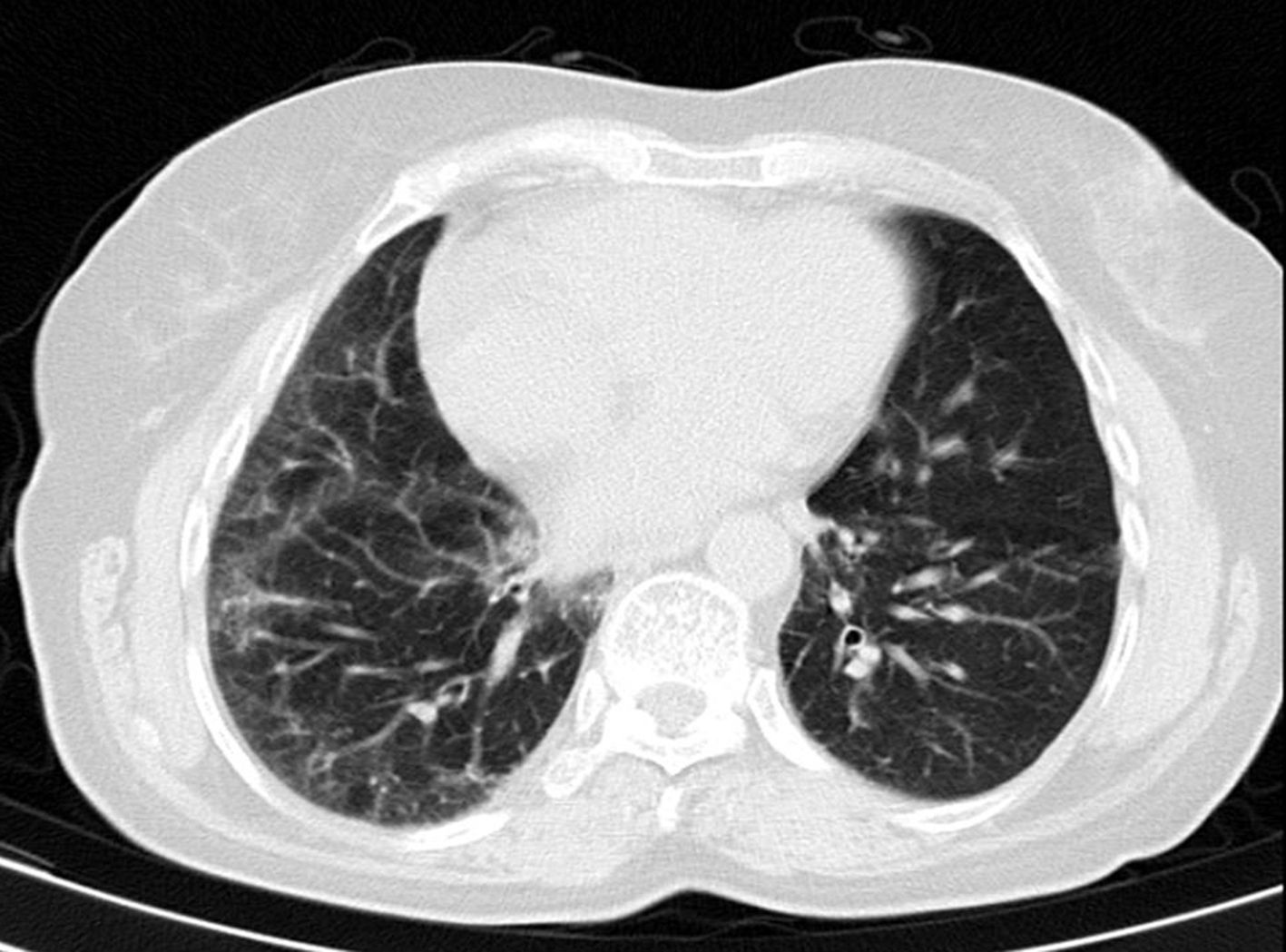
Mujer de 76 años, con antecedentes de trombosis venosa profunda idiopática derecha en 2006, osteoporosis posmenopáusica con aplastamientos vertebrales dorsales, poliartrosis generalizada y reciente diagnóstico de síndrome depresivo. Sigue tratamiento con paracetamol 1g/8h, escitalopram 20mg/24h, risedronato 35mg/semanal y carbonato cálcico 2.500mg/colecalciferol 880U/24h. Remitida a consulta desde atención primaria por presentar clínica de astenia, malestar general y poliartralgias generalizadas atribuidas a artrosis. No presenta focalidad clínica ni cuadro constitucional asociado, a excepción de disnea a grandes esfuerzos, realizando despistaje analítico general incluyendo perfil tiroideo sin apreciar alteraciones; se le diagnostica de síndrome depresivo sin respuesta a tratamiento. En la exploración destaca hipercifosis dorsal, crepitantes pulmonar basal derecho a la auscultación, y manos con dedos hiperqueratósicos y fisuras laterales de carácter bilateral «manos de mecánico». La paciente refiere esta afectación dermatológica junto con palidez-cianosis de dedos en relación al frío, de 6 años de evolución, compatible con el fenómeno de Raynaud. El resto de la exploración general es normal. Valoración geriátrica integral: a) escala de Lawton-Brody para actividades instrumentales: 6; b) test de Pfeiffer: 1; c) escala de Yasevage: 12, y d) escala de Gijón: 9. El estudio analítico presenta un hemograma y hemostasia normal. Perfil renal, hepático, lipídico y tiroideo normal. CPK y aldolasa normales, ANA positivo 1/80 moteado, anti-Ro/SSA y anti-Jo positivo (identificado por técnica de ELISA). ANCA, inmunoglobulinas, FR, péptido citrulinado, proteinograma, complemento, anticardiolipinas, anticoagulante lúpico, virus de la hepatitis C y reacción de Mantoux, normales o negativos. Sedimento urinario sin alteraciones. Destacaba una PCR de 1,4mg/dl (N<0,3mg/dl) y VSG de 54mm la 1.ª hora. La radiografía de tórax muestra un patrón intersticial fino basal derecho, las radiografías de ambas manos únicamente mostraban signos artrósicos. El electrocardiograma no muestra alteraciones, y la ecocardiografía describe una insuficiencia tricúspide leve y PSAP de 32mmHg. El TAC de alta resolución de tórax señala un infiltrado intersticial inespecífico leve en base derecha sin otros hallazgos (fig. 1) y el TAC de abdomen-pelvis no aprecia enfermedad. Las pruebas funcionales respiratorias demuestran un patrón restrictivo leve sin afectación de la difusión. La electromiografía resulta normal y en la capilaroscopia, la presencia de escasos megacapilares, así como afilamiento de la vascularización en ambas manos. Con estos datos se diagnostica a la paciente de síndrome antisintetasa sin compromiso muscular, iniciándose tratamiento con deflazacort 30mg/24h en pauta descendente dada la incipiente afectación pulmonar, sin asociar inmunosupresores por negativa de la paciente. Tras 12 meses de seguimiento la paciente presenta remisión clínica, radiológica e inflamatoria (PCR y VSG), mejorando incluso levemente la hiperqueratosis y fisuras de las manos; actualmente se encuentra libre de esteroides y sin datos de afectación muscular en el seguimiento.

Las enfermedades autoinmunes sistémicas son afecciones infrecuentes con aumento de su prevalencia en el paciente anciano debido al aumento en la supervivencia, y como consecuencia de la inmunosenescencia del sistema inmune. Se estima que afectan a un 20% de los ancianos siendo las enfermedades más frecuentes entre otras el síndrome de Sjögren, la polimialgia reumática/arteritis de células gigantes, la artritis reumatoide y las miopatías inflamatorias idiopáticas1,2.
El SA es una entidad muy infrecuente asociada habitualmente a las miopatías inflamatorias idiopáticas. Existen publicados unos criterios propuestos no validados, además de los clásicos de Bohan y Peter3, en los que ante la presencia de anti-Jo-1 (o anti-PL7,anti-PL12, anti-EJ, anti-OJ) y una manifestación como neumopatía intersticial, miositis, alteraciones capilaroscópicas, artritis, «manos de mecánico», fenómeno de Raynaud, disfunción esofágica o calcinosis, se acepta el diagnóstico de SA4. La afectación pulmonar intersticial es el rasgo clínico más relevante del SA por su potencial impacto en el pronóstico y supervivencia, siendo poco frecuente que dicha afección preceda a la miositis, la cual puede aparecer años después, o cursar de manera subclínica; siendo infrecuente (hasta un 20% de los casos) aunque descrita la ausencia de dicha afección muscular en este síndrome5. Los anti-Jo-1 son anticuerpos más específicos para la afectación muscular y pulmonar, aunque su presencia no presenta asociación a una mayor tasa de recurrencias o peor pronóstico, relacionándose su presencia incluso a formas más benignas de neumopatía intersticial6. La presencia de anticuerpos antisintetasa diferentes del anti-Jo-1 (no Jo-1) suelen asociarse con mayor frecuencia a neumopatía intersticial con escasa o inexistente afectación muscular7.
El tratamiento del SA debe ser adecuado a la severidad, compromiso pulmonar y muscular, así como a las circunstancias del paciente, especialmente en el anciano. El pilar básico es la terapia esteroidea asociada a inmunosupresores como ciclofosfamida, azatioprina, metotrexato, ciclosporina; describiéndose buenos resultados con el empleo de inmunoglobulinas, micofenolato mofetilo, anti-TNF o rituximab en los casos más severos8. El pronóstico y la supervivencia de los pacientes con SA es poco conocida por el escaso número de pacientes en las series publicadas, quedando condicionado principalmente a la afectación pulmonar intersticial9. La remisión únicamente con tratamiento esteroideo es alcanzada en el 25-68% de los casos, siendo esta difícil de mantener incluso con la asociación de inmunosupresores10.
El caso que exponemos representa una forma de presentación y evolución excepcional tanto por la ausencia de afectación muscular, como por la buena evolución clínica con el empleo de terapia simplificada a baja intensidad.
Por lo tanto, dado el aumento de la prevalencia de las enfermedades autoinmunes sistémicas en el paciente anciano, creemos que es necesario ampliar las sospechas diagnósticas a este grupo de enfermedades, difíciles de identificar por el profesional inexperto, así como por presentar manifestaciones clínicas habitualmente inespecíficas y compartidas por múltiples enfermedades más prevalentes. De este modo podremos realizar una adecuada, temprana y adaptada intervención terapéutica tras una valoración geriátrica integral, en pro de la mejora en el pronóstico, así como en la calidad de vida en el paciente de edad avanzada.
