




La actual “epidemia de obesidad”, y su correlato, el síndrome metabólico, se ha puesto en relación en los últimos años no solamente con enfermedad cardiovascular, sino con la presencia de múltiples enfermedades crónicas, e incluso con el desarrollo de incapacidad. Por ello, se ha llegado a establecer la hipótesis de que esta situación de riesgo y la situación de fragilidad que tanto interés ha despertado en el ámbito de la Geriatría compartirían mecanismos fisiopatológicos comunes. Estos mecanismos constituyen el objeto de esta revisión.
In recent years, the current “obesity epidemic” and its correlate, metabolic syndrome, have been related not only to cardiovascular disease but also to the presence of multiple chronic diseases and even to the development of disability. Therefore, it has been hypothesized that this situation of risk and that of frailty, which has aroused such interest in geriatrics, could share common physiopathologic mechanisms. These mechanisms are reviewed in the present article.
La costumbre, lo cotidiano, aquello que nos parece obvio y bien establecido nos impide en ocasiones profundizar en las posibles causas de los fenómenos observables.
Damos por conocido y consideramos incuestionable que la enfermedad en el anciano presenta una serie de características especiales1, tan decisivas para el manejo médico de los problemas de salud de esta población, que hacemos de ellas la base de nuestra especialidad, nuestra razón de ser.
La cronicidad, la tendencia a la incapacidad, la comorbilidad, la variabilidad y la atipicidad en la presentación de la enfermedad son los principales aspectos diferenciales que aducimos para justificar nuestra existencia2.
Son ya numerosas las series que establecen una incidencia de deterioro funcional secundario a la hospitalización de alrededor de un 25% de los casos (al mes del alta hospitalaria), incapacidad que se “fija” a partir de este punto temporal, permaneciendo estable a los tres meses de evolución3. La incapacidad justifica algunas variaciones en los resultados asistenciales que deben ser tenidas en cuenta en las comparaciones entre los servicios y las organizaciones asistenciales4.
¿Cuál es el substrato fisiopatológico que subyace a las manifestaciones propias de la enfermedad crónica y a su potencial incapacitante?
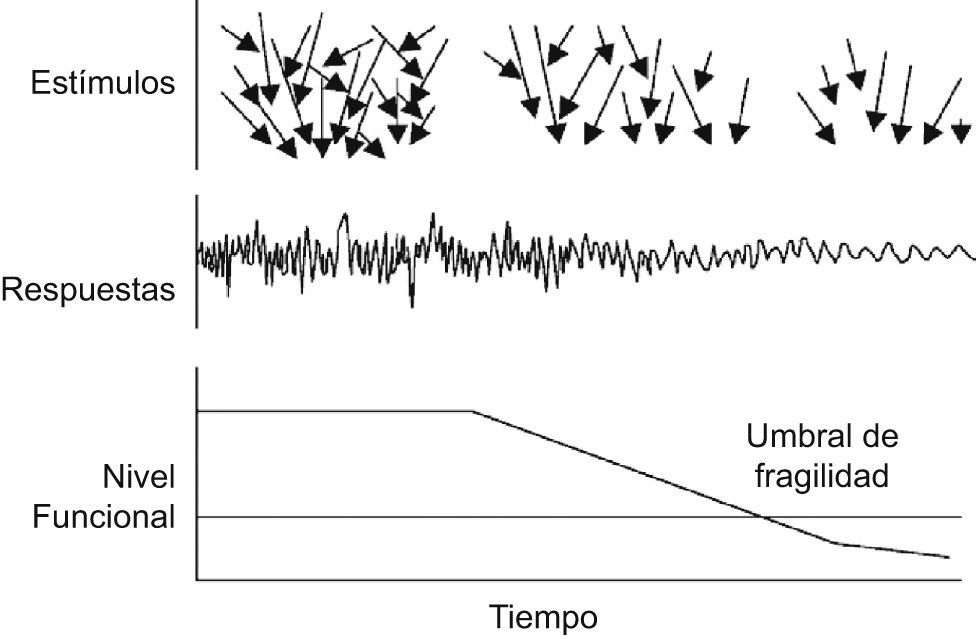
Concepto de fragilidadSe han realizado grandes avances en este ámbito en la última década. Especialmente, el concepto de fragilidad ha permitido consensuar características que asociadas desembocan en una vía final común, la pobre respuesta al estrés en una elevada proporción de la población anciana5–8. La amplitud de ésta se va reduciendo, situando a estos individuos en una situación en la que la posibilidad de acceso a los estímulos es cada vez menor, así como el rango de su respuesta, situación que se retroalimenta a lo largo del tiempo, generando una progresiva reducción de su rendimiento funcional9,10 (fig. 1).

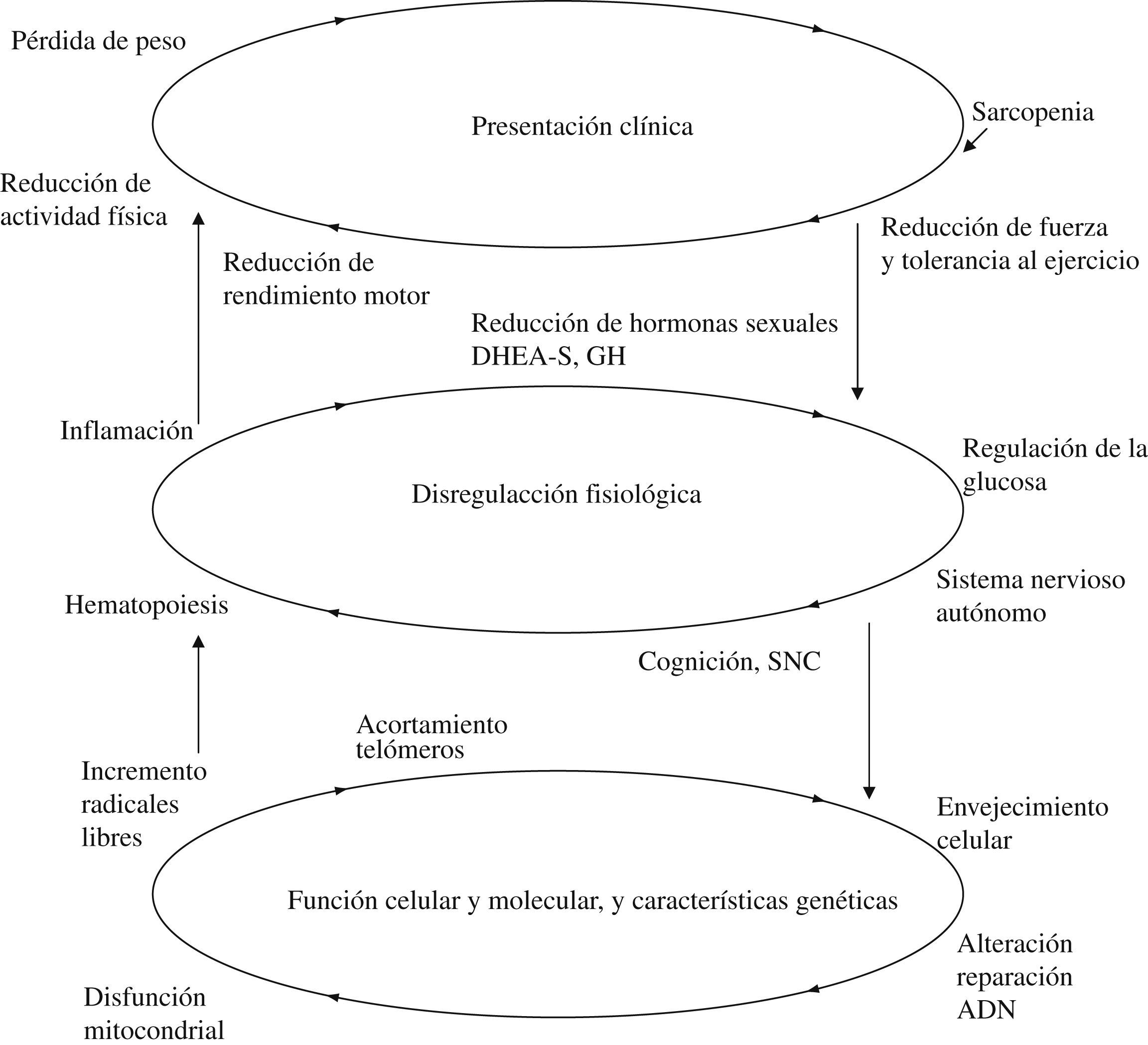
En este marco conceptual se han descrito fenotipos relacionados con esa reducción del rendimiento, así como determinados componentes subclínicos, mecanismos fisiológicos y relaciones entre genética y entorno que se reflejan en un esquema ya clásico descrito por Fried et al (fig. 2)11,12.

Entre los factores implicados en el modelo representado en la figura 2, han recibido especial atención en los últimos años la reducción de rendimiento motor (disminución de fuerza y tolerancia al ejercicio y su correlato: la sarcopenia) desde el punto de vista clínico, la inflamación y la alteración del metabolismo de la glucosa desde la perspectiva fisiopatológica y la disfunción mitocondrial y la generación de radicales libres desde la Biología Celular13–18. Todos ellos se encontrarían interrelacionados, de modo que la afectación de uno de los factores en equilibrio daría lugar a manifestaciones detectables en los restantes ámbitos.
Por ello, el abordaje de la enfermedad, y de su correlato, la funcionalidad (el rendimiento), en el paciente anciano debe partir siempre de una perspectiva sistémica, en la que se ven implicados tanto los órganos efectores (los músculos, el cerebro) como los órganos reguladores (el páncreas, de especial interés en el tema que nos va a ocupar a continuación) a través de modos de respuesta subcelulares (generación de radicales libres y mediadores inflamatorios) presentes en todos los tejidos (sistémicos).
Síndrome metabólico, ¿modelo de envejecimiento acelerado?Para nuestro grupo de investigación, este esquema se ve reproducido en muchos aspectos en una situación de riesgo atractiva para los investigadores desde hace años: el síndrome metabólico (SM). Estas relaciones serán el motivo de esta reflexión, que plantea la posibilidad de considerar este síndrome como un modelo de envejecimiento acelerado.
El SM es un constructo basado en hallazgos epidemiológicos que plantean que la combinación de algunos factores de riesgo tiene un efecto final multiplicativo respecto a cada uno de ellos considerado individualmente19–24, lo que debería justificarse por un trasfondo fisiopatológico explicativo.
La combinación de obesidad, hipertensión, hipercolesterolemia y diabetes aparece en todas las definiciones (International Diabetes Foundation [IDF], National Cholesterol Education program. Adult treatment panel III [NCEP ATP III], Organización Mundial de la Salud [WHO], American Association of Clinical Endocrinologists [AACE])25. Sin embargo, los puntos de corte considerados en cada caso son diferentes. Ello se justifica por las divergentes consideraciones realizadas en cada caso respecto a la relación riesgo-beneficio del tratamiento.
Por otra parte, en algunos casos se incluyen en el síndrome factores de riesgo emergentes, para los que existe un menor consenso (presencia de marcadores bioquímicos de inflamación o de estado protrombótico, por ejemplo)26,27.
Manteniéndonos en el terreno de las asociaciones epidemiológicas, el SM se ha puesto en relación con el riesgo de desarrollo de insuficiencia cardíaca, apnea del sueño, fractura de la cadera, cáncer endometrial, síndrome depresivo y deterioro cognitivo, disfunción ventilatoria restrictiva, trombosis venosa, osteoporosis, reducción de la fuerza muscular y, lo que es aún más interesante para nosotros, aparición de incapacidad28–41.
Creo que podríamos suscribir que todas estas condiciones, y en especial su asociación y relación con la probabilidad de desarrollo de deterioro funcional, son habituales en el paciente anciano, y en especial en el paciente anciano frágil.
¿Podemos describir semejanzas entre la fisiopatología del SM y la fisiología del envejecimiento?, ¿puede constituir aquél un modelo de éste? Desde nuestro punto de vista la respuesta es afirmativa.
Así, desde un punto de vista genético, las laminopatías (enfermedades genéticas que se asocian a un amplio espectro de fenotipos y son debidas a mutaciones del gen que codifica la lámina nuclear A/C) afectan el sistema cardiovascular a través de una vía clave: la aterosclerosis. En una de ellas, la lipodistrofia familiar parcial del tipo Dunnigan, la aterosclerosis se relaciona con dislipidemia, hiperinsulinemia, hipertensión y diabetes (SM), mientras que en la progeria —síndrome de Hutchinson-Gilford— la aterosclerosis se desarrolla con una exposición menor a estos factores, reflejando el proceso de envejecimiento acelerado que la caracteriza. En ambos casos, las enfermedades secundarias y relacionadas con la aterosclerosis constituyen la causa principal de mortalidad42.
En ambos casos, se aprecia un incremento proporcional de tejido graso corporal y una distribución atípica de éste, con predominio visceral y especialmente perivascular43,44. Estas dos últimas localizaciones son especialmente activas desde el punto de vista metabólico.
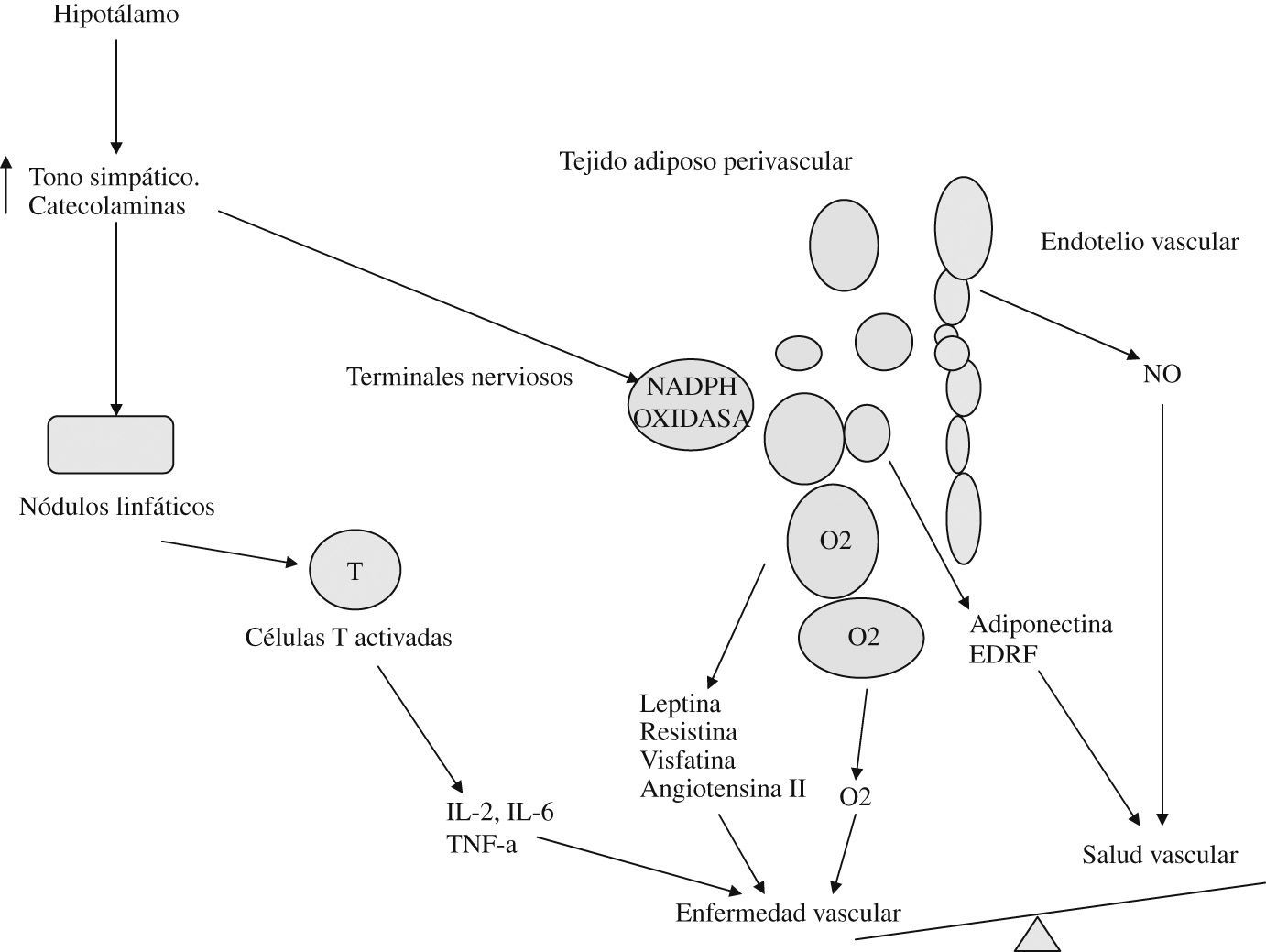
Tejido grasoUna aproximación a la actividad del tejido adiposo perivascular se refleja en la figura 345.

Como vemos, el adipocito es capaz de sintetizar varias moléculas con capacidad de actuar directa o indirectamente sobre la pared vascular, influyendo en el equilibrio entre salud y enfermedad vascular.
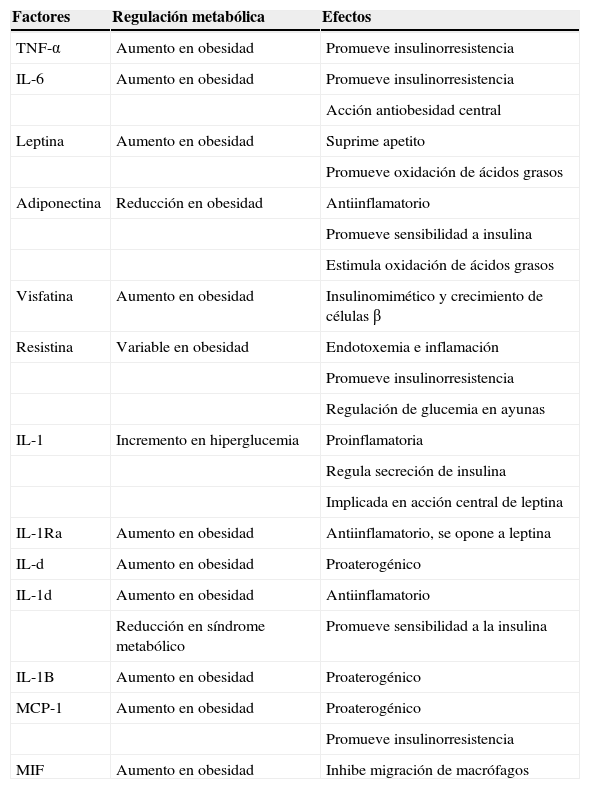
Su actividad parece relacionarse con la situación oxidativa a nivel local (generación de especies reactivas de oxígeno) y la presencia de mediadores inflamatorios (citoquinas). Se puede hablar incluso de una intersección entre metabolismo e inmunidad, de modo que macrófago y adipocito comparten algunas funciones (tabla 1)45–52.
Factores que median la intersección entre metabolismo e inmunidad
| Factores | Regulación metabólica | Efectos |
| TNF-α | Aumento en obesidad | Promueve insulinorresistencia |
| IL-6 | Aumento en obesidad | Promueve insulinorresistencia |
| Acción antiobesidad central | ||
| Leptina | Aumento en obesidad | Suprime apetito |
| Promueve oxidación de ácidos grasos | ||
| Adiponectina | Reducción en obesidad | Antiinflamatorio |
| Promueve sensibilidad a insulina | ||
| Estimula oxidación de ácidos grasos | ||
| Visfatina | Aumento en obesidad | Insulinomimético y crecimiento de células β |
| Resistina | Variable en obesidad | Endotoxemia e inflamación |
| Promueve insulinorresistencia | ||
| Regulación de glucemia en ayunas | ||
| IL-1 | Incremento en hiperglucemia | Proinflamatoria |
| Regula secreción de insulina | ||
| Implicada en acción central de leptina | ||
| IL-1Ra | Aumento en obesidad | Antiinflamatorio, se opone a leptina |
| IL-d | Aumento en obesidad | Proaterogénico |
| IL-1d | Aumento en obesidad | Antiinflamatorio |
| Reducción en síndrome metabólico | Promueve sensibilidad a la insulina | |
| IL-1B | Aumento en obesidad | Proaterogénico |
| MCP-1 | Aumento en obesidad | Proaterogénico |
| Promueve insulinorresistencia | ||
| MIF | Aumento en obesidad | Inhibe migración de macrófagos |
IL: interleukina; IL-1Ra: antagonista del receptor de interleukina 1; MCP-1: proteína quimiotáctica del monocito; MIF: factor inhibidor de la migración del macrófago; TNF-α: factor de necrosis tumoral alfa.
Entre las sustancias implicadas en este estatus fisiológico, podemos destacar como más relevantes, según los conocimientos actuales, las siguientes:
- •
la adiponectina;
- •
la leptina;
- •
la resistina;
- •
la visfatina;
- •
el TNF-α;
- •
la interleukina (IL) 6,
- •
y la proteína C reactiva53–60.
La adiponectina incrementa la vasodilatación endotelio dependiente, suprimiendo el proceso aterosclerótico, reduciendo los efectos y niveles de TNF y atenuando los efectos del factor de crecimiento en el músculo liso vascular. Asimismo, inhibe los efectos endoteliales de las lipoproteínas de baja densidad (LDL, low density lipoproteins) oxidados, incluyendo la supresión de la generación de superóxido. También estimula la angiogénesis, reduce el engrosamiento de la íntima limitando la proliferación de las células musculares lisas en las arterias con daño mecánico.
Se reconoce como una red de señales entre adipocitos, tejidos sensibles a la insulina y función vascular, con implicaciones en el riesgo cardiovascular, frenando el desarrollo de fibrosis hepática, actuando como hormona antiinflamatoria (regula a la baja la secreción de adipokinas), reduciendo el contenido graso del músculo y consiguiendo una reducción de glucemia independiente de insulina56,59.
La leptina, por su parte, regula el apetito y el balance de energía mediante una acción central (hipotálamo), por lo que se la ha considerado como la hormona de la saciedad. Sus niveles están condicionados por la masa grasa corporal53.
La resistina puede causar resistencia a la insulina, ligando obesidad y diabetes mellitus tipo ii. Incrementa la glucemia y la insulinemia y sus efectos sobre el adipocito conllevan la liberación de otras moléculas prodiabéticas (ácidos grasos no esterificados), especialmente en ancianos. Se considera igualmente un marcador proinflamatorio57,58.
Este “foco” sobre el adipocito (célula de especial relevancia en obesos y ancianos) pone de manifiesto los “puntos clave” sobre los que se asienta buena parte de la fisiopatología común del SM y el envejecimiento:
- •
estado proinflamatorio (IL, TNF-α, proteína C reactiva [PCR]);
- •
situación oxidativa (acción antioxidante de la adiponectina);
- •
resistencia a la insulina (resistina, visfatina),
- •
e hiperlipidemia (resistina).
Aunque en este listado hemos intentado poner en relación cada molécula con una determinada acción, lo realmente importante en el resultado metabólico final es la relación entre todas ellas, que ha dado lugar al concepto de metaboloma.
El metaboloma se refiere a todos los elementos de bajo peso molecular de un sistema biológico (intermediarios metabólicos, hormonas, moléculas señal) y a sus interrelaciones, y refleja las consecuencias integradas del genotipo, la conducta y el ambiente61.
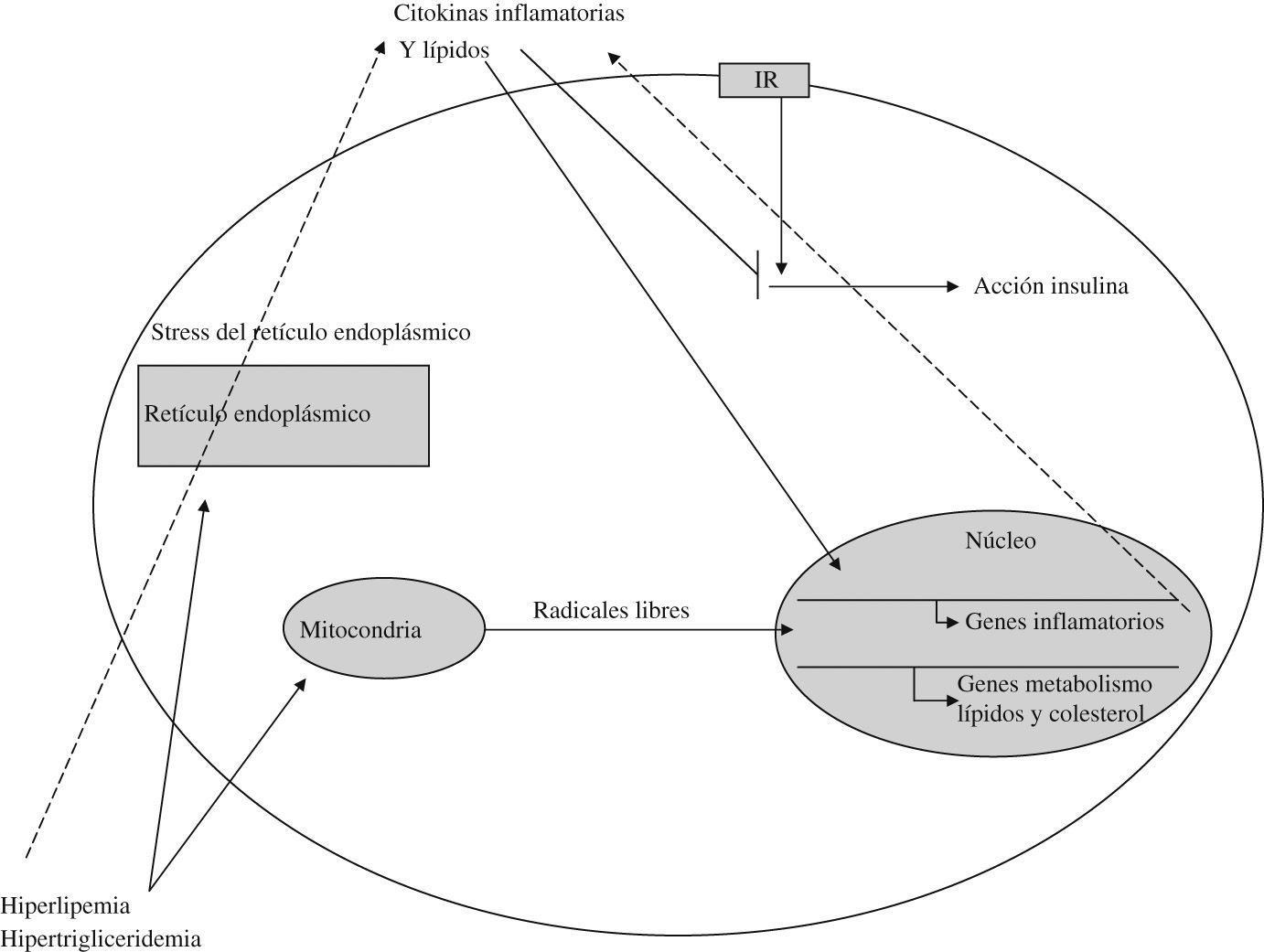
A nivel subcelular, estos elementos actúan fundamentalmente sobre la mitocondria y el retículo endoplásmico, según se refleja en la figura 462.

Hiperlipidemia, hiperglucemia y citokinas inflamatorias propician la generación mitocondrial de radicales libres y el estrés del retículo endoplásmico62,63, que a través de las vías metabólicas señaladas dan lugar a una mayor resistencia a la insulina y a la activación de genes inflamatorios y del metabolismo lipídico, cerrando el círculo de alimentación del sistema.
Páncreas en el síndrome metabólicoHablar de SM es referirse, como sabemos, a una situación de disregulación del metabolismo de la glucosa. En ella, la célula betapancreática es tanto un órgano efector como afectado por la situación fisiológica creada.
Una serie de moduladores primarios (hiperglucemia, hiperlipidemia, leptina y citokinas) y secundarios (autoinmunidad y drogas) actuan sobre factores locales:
- •
masa predeterminada de células β;
- •
sensibilidad a señales proapoptóticas;
- •
potencial regenerativo de las células β;
- •
citokinas derivadas de los islotes (IL-6, TNF-α);
- •
moléculas señal (factor de transcripción nuclear Kappa B [NF-KB], disfunción mitocondrial, estrés oxidativo, estrés del retículo endoplásmico),
- •
y amiloide
y dan lugar al resultado funcional final observable64–70.
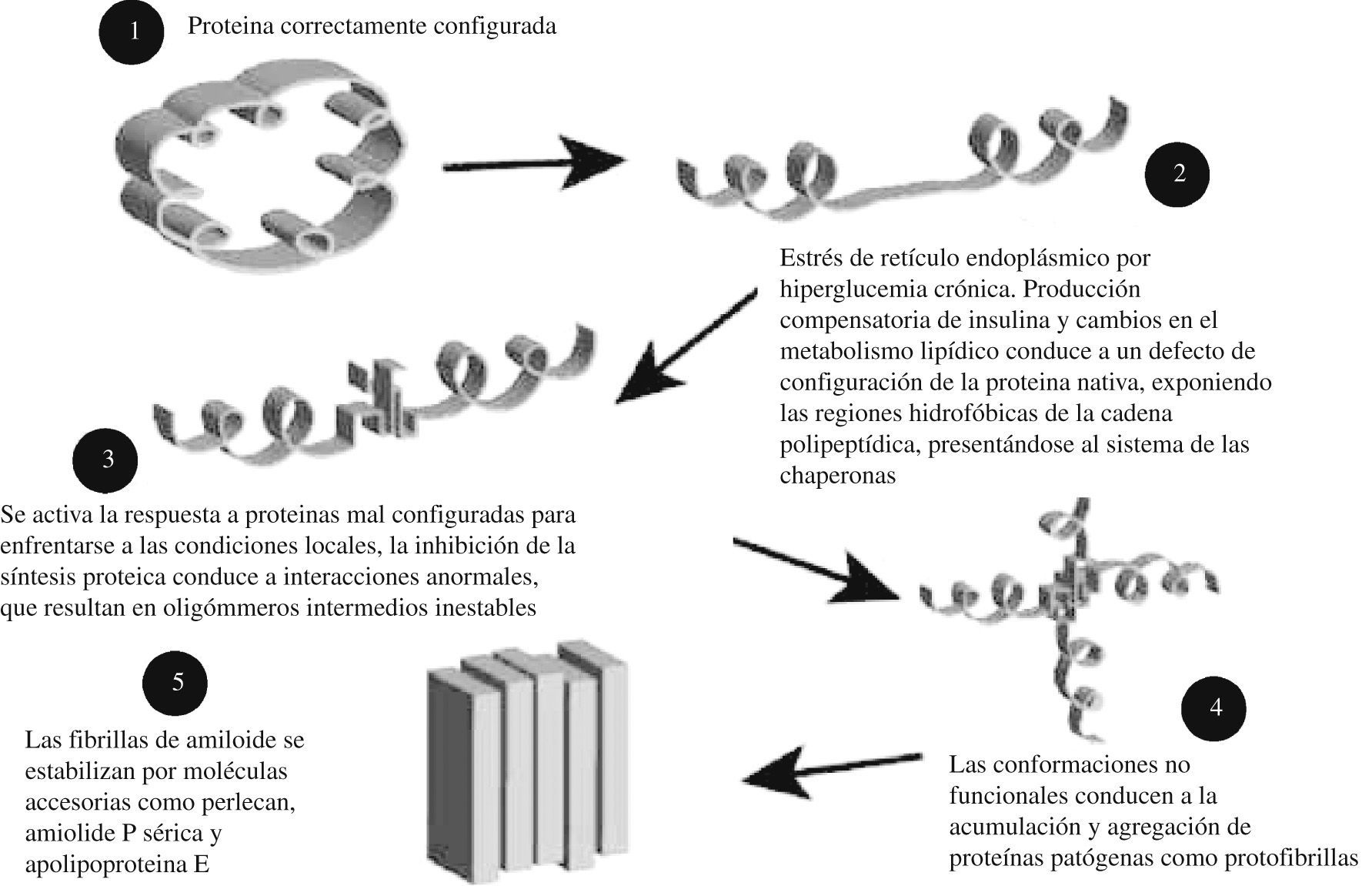
Su interacción en el SM origina una de las llamadas “enfermedades conformacionales” (como la enfermedad de Alzheimer, en la que se aducen algunos mecanismos similares, y que también se ha puesto en relación con el SM) por depósito de proteínas anormalmente constituidas, en concreto, la denominada polipéptido amiloide del islote68,69.
El mecanismo por el que se lleva a cabo este depósito se muestra en la figura, y depende de la imposibilidad de contrarrestar la situación de estrés a nivel del retículo endoplásmico, secundaria a la aparición de proteínas mal configuradas que supera la “respuesta a proteínas mal configuradas” (unfolded protein response) del retículo endoplásmico71–75 (fig. 5).

Esta respuesta a proteínas mal configuradas genera una defensa antioxidante, la supresión de la síntesis de proteínas, regulación al alza de las chaperonas (estabilizan las proteínas protegiendo su configuración) y ubiquitina (degradación de proteínas mal configuradas) y secundariamente inflamación y apoptosis, condicionando una reducción de la secreción insulínica (menor reserva funcional pancreática)69.
Función muscular en el síndrome metabólicoLa célula muscular, como consecuencia de su actividad, también da lugar a la producción de citokinas inflamatorias (IL-6, TNF), cerrando como órgano efector el círculo inflamatorio en el SM y el envejecimiento, que se refleja en la reducción del rendimiento final observable76–79.
Dicha liberación es dependiente del tipo, la intensidad y la duración del ejercicio79.
Esta expresión de citokinas inflamatorias se ve reducida mediante el progresivo entrenamiento de moderada intensidad79.
La combinación de predominio adiposo y reducción de masa y disfunción muscular inflamatoria resulta tan disruptiva desde el punto de vista funcional que ha dado origen al concepto de “obesidad sarcopénica”, que debemos considerar como una situación de decondicionamiento fisiológico extremo que pone al sujeto en elevado riesgo de incapacidad.
Dieta (reducción del tejido adiposo) y ejercicio aeróbico (reacondicionamiento muscular que reduce la situación inflamatoria durante el desarrollo de la actividad de un sujeto concreto) se constituyen, pues, en pilares del tratamiento de este complejo fisiológico.
Influencia de la hipoxiaAl comentar el comportamiento del órgano efector (el músculo) hemos hecho referencia a la importancia de su reacondicionamiento mediante el trabajo en situación aeróbica.
Ello es debido a que la producción de radicales libres presenta una distribución bimodal, de forma que en situaciones de hipoxia e hiperoxia se produce un aumento en la formación de éstos80–82.
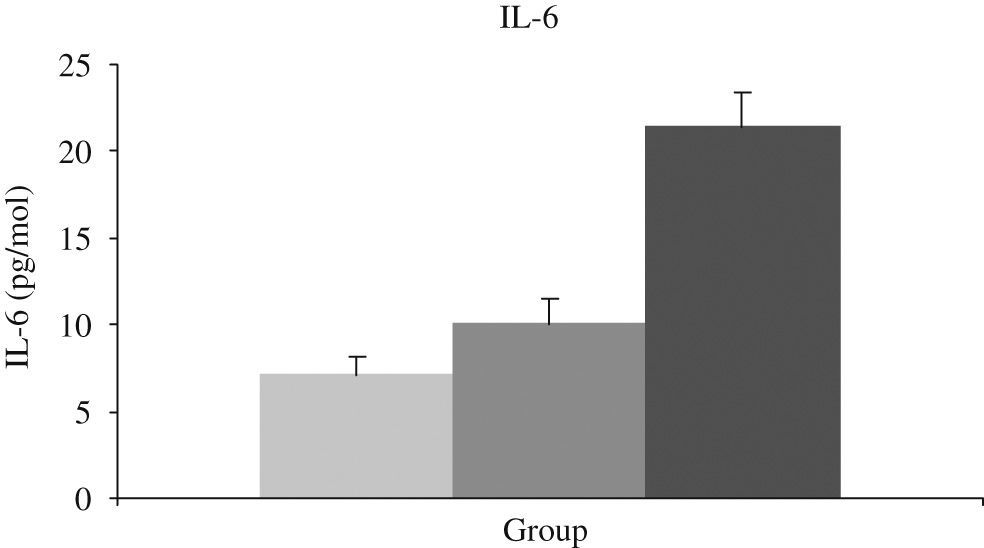
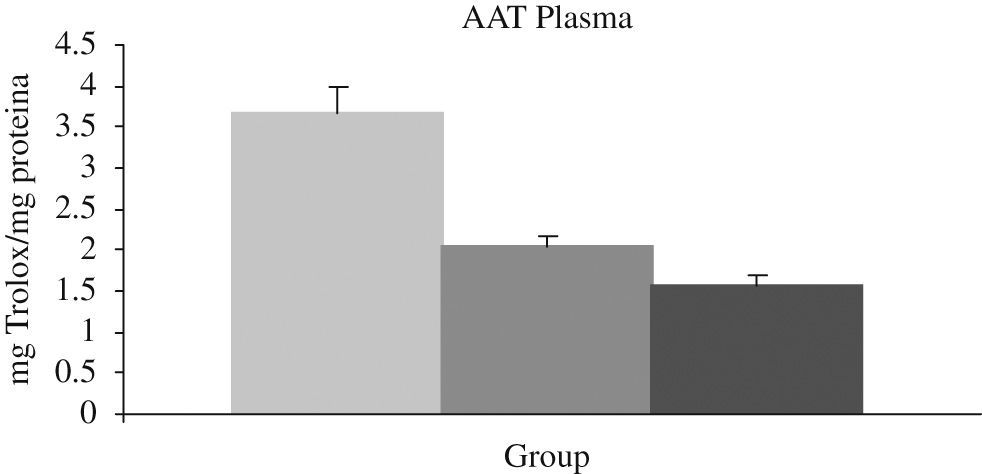
Este hecho ha sido comprobado en población anciana enferma por nuestro grupo, detectando una menor actividad antioxidante en situaciones de hipoxia y una mayor actividad inflamatoria (IL-6) en sangre durante ésta (figs. 6 y 7).
.")
.")
Así, aquellas situaciones que ponen al sujeto en situación de riesgo de ejecutar función en situaciones de hipoxia (insuficiencia cardíaca [IC], enfermedad pulmonar obstructiva crónica [EPOC]) deberían revelar un elevado potencial incapacitante, hecho que comprobamos diariamente en nuestro trabajo clínico, ya que alimentarían la continua generación de radicales libres, inflamación y deterioro funcional.
ConclusiónLa asociación entre SM y enfermedad crónica es un hecho incuestionable desde el punto de vista epidemiológico. Como se ha comentado inicialmente, estas enfermedades son habituales en el anciano, en el que han demostrado un elevado potencial incapacitante.
Por otra parte, en todas ellas, al estudiar los posibles mecanismos fisiopatológicos subyacentes se aduce un papel para los factores mencionados en este apartado. Además, las más relevantes, entre ellas IC y EPOC, cursan con hipoxia, y en el caso del anciano se asientan sobre un terreno en el que predomina la grasa visceral, muy activa metabólicamente, como hemos visto.
Las interacciones entre las moléculas implicadas en estas vías metabólicas deben, sin duda, formar parte de la fisiopatología de la cronicidad y del potencial incapacitante de la enfermedad crónica, abriendo vías para la comprensión del concepto de fragilidad.
