




Extraskeletal myxoid chondrosarcoma is a rare soft tissue tumour with a high local and distant metastasis rate and limited response to chemotherapy.
Meckel's diverticulum is the most frequent congenital anomaly, and it is associated with a considerable risk of malignant transformation.
In this case report, we describe a 50-year-old female patient with a history of extraskeletal myxoid chondrosarcoma of the lower limb and metastasis to the forearm who went to the emergency department with abdominal pain. The investigations revealed a caecal volvulus. A lesion in the middle third of the ileum was incidentally discovered and removed during surgery.
Pathology examination revealed a Meckel's diverticulum adenocarcinoma, with metastasis of extraskeletal myxoid chondrosarcoma.
Resection was complete; however, the patient had diffuse metastatic pulmonary disease and died eight months later due to disease progression.
This mechanism of tumour-to-tumour metastasis is described in other locations, but, regarding the Meckel's diverticulum, this is a unique situation, previously unreported in the literature.
El condrosarcoma mixoide extraesquelético es un tumor de tejidos blandos poco frecuente, con una elevada tasa de recurrencia y metástasis a distancia y una respuesta limitada a la quimioterapia.
El divertículo de Meckel es la anomalía congénita más frecuente y se asocia a un riesgo considerable de transformación maligna.
En este caso clínico describimos a una paciente de 50 años con antecedentes de condrosarcoma mixoide extraesquelético de miembro inferior y metástasis en el antebrazo que acudió al servicio de urgencias por dolor abdominal. La exploración reveló un vólvulo cecal. Se descubrió incidentalmente una lesión en el tercio medio del íleon, que se extirpó durante la intervención quirúrgica.
El examen patológico reveló un adenocarcinoma de divertículo de Meckel, afectado por metástasis de condrosarcoma mixoide extraesquelético.
La resección fue completa; sin embargo, la paciente presentaba enfermedad pulmonar metastásica difusa y falleció ocho meses después debido a la progresión de la enfermedad.
Este mecanismo de metástasis entre tumores está descrito en otras localizaciones, pero en lo que respecta al divertículo de Meckel, se trata de una situación única en la literatura.
Extraskeletal myxoid chondrosarcoma (EMC) is a sporadic subtype of sarcoma, usually arising in the extremities and classified by the World Health Organization classification as a tumour of uncertain differentiation.1 EMC is usually diagnosed in middle age, it has an indolent course exhibiting a propensity for local recurrence and metastasis.2
Meckel's diverticulum (MD) is the most frequent congenital anomaly of the gastrointestinal tract3 and is considered a high-risk area for malignant transformation, most commonly neuroendocrine tumours.3,4 This risk increases with age, with a reported incidence of up to 3.2%.5
Tumour-to-tumour metastasis is a rare phenomenon in which primary tumour cells metastasize into another tumour. It is mainly attributed to the tumour's higher vascularization, which will harbour other tumour cells, creating a favourable niche for tumour growth.6
Albeit rare, it is described in some organs, namely the thyroid7 and ovary,8 but there is no description in the literature of a tumour-to-tumour to a Meckel's diverticulum adenocarcinoma.
In this report, we aim to describe a metastasis of an EMC to an adenocarcinoma arising in a MD, constituting a type of tumour-to-tumour metastasis which, as far as the authors know, has not been reported in the literature.
Case reportA 50-year-old female patient with a history of amputation of the right lower leg five years earlier due to EMC and metastases to the right forearm in the previous year presented to the emergency department with abdominal pain. The imaging workup revealed a caecal volvulus. During surgery, there was an incidental finding of a lesion in the midgut. A right hemicolectomy with small bowel resection was performed.
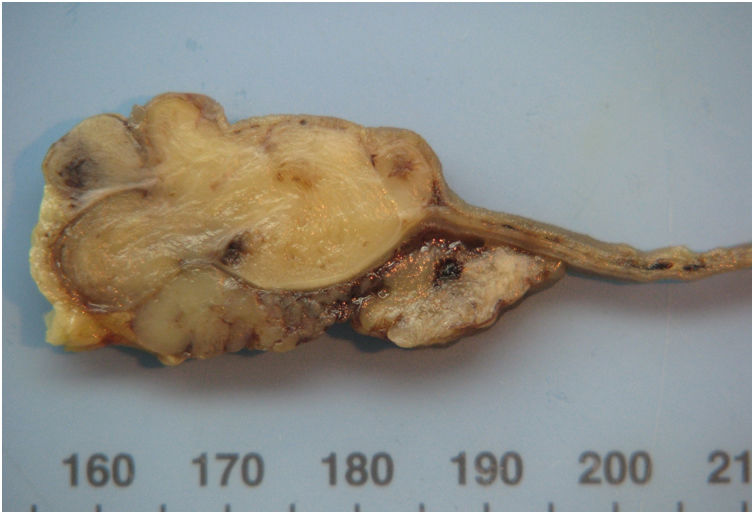
The right hemicolectomy revealed an ischaemic lesion in association with the caecal volvulus. The small bowel exhibited a 4cm ulcerated lesion in the mucosa, with central umbilication. On the cut section, it was composed of grey and lobular cartilaginous-like tissue surrounding a small slit (Fig. 1).
.")
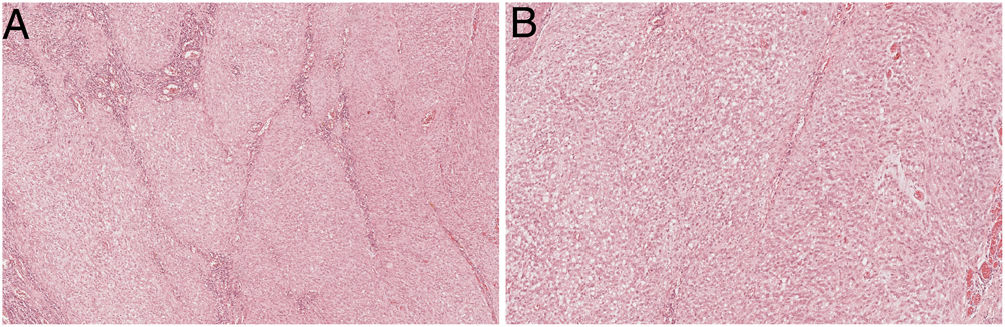
Histologically, the lesion was composed of oval cells in a multinodular pattern, embedded in a myxoid matrix without evident chondroid differentiation (Fig. 2A and B). Some areas had a trabecular pattern, and ischaemic necrosis was present. The tumour was intraparietal with serosal and submucosal involvement.
 The lesion had a multinodular pattern with a mild myxoid background and necrosis (upper left), H&E 20×. (B) In the most cellular areas, the cells have a more undifferentiated phenotype, with pronounced atypia and irregular nuclear membrane with visible nucleoli, H&E 200×.")
Extraskeletal myxoid chondrosarcoma. (A) The lesion had a multinodular pattern with a mild myxoid background and necrosis (upper left), H&E 20×. (B) In the most cellular areas, the cells have a more undifferentiated phenotype, with pronounced atypia and irregular nuclear membrane with visible nucleoli, H&E 200×.
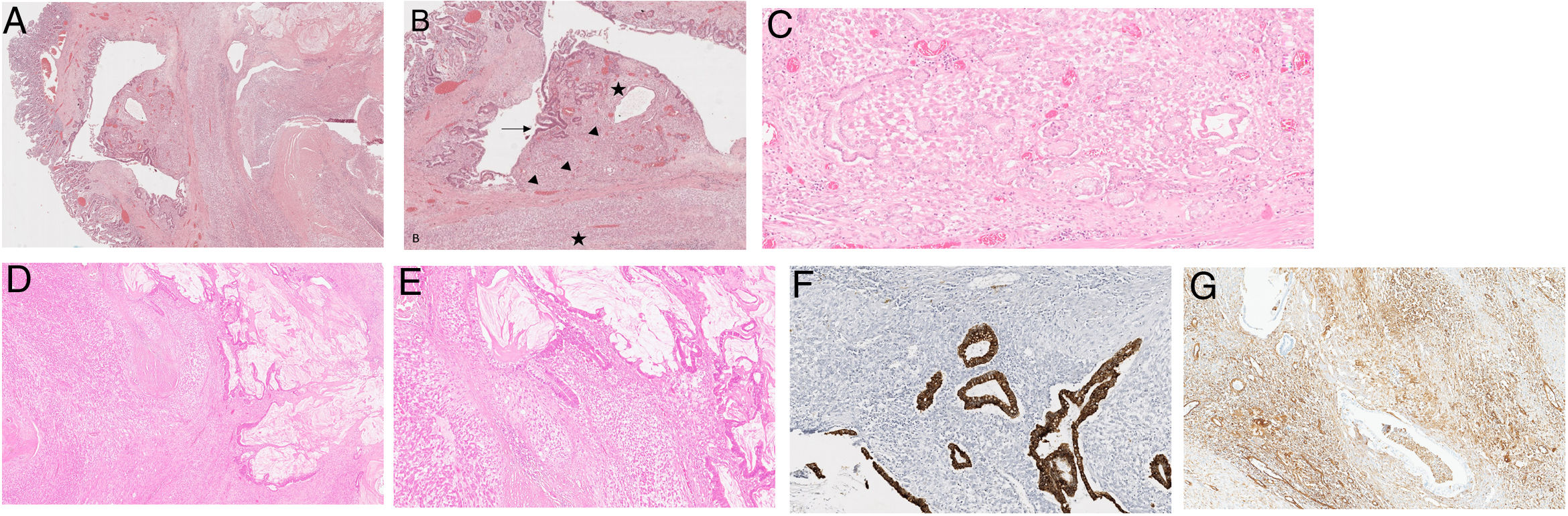
The slit revealed to be a diverticulum with small bowel, gastric (Fig. 3C), and pancreatic mucosa, associated with low and high-grade dysplasia (Fig. 3A and B), as well as invasive adenocarcinoma with mucin production (Fig. 3D and E) that infiltrated the adipose tissue. The myxoid neoplasm diffusely surrounded the diverticulum.
 The slit corresponded to a diverticulum mainly with ileal type mucosa lining, H&E20×. (b) Some heterotopic gastric type glands were also present (arrowheads) and foci of dysplasia (arrow). The majority of the diverticulum was affected by the extraskeletal myxoid chondrosarcoma (EMC) (star). (c) (Inlet) – heterotopic gastric type glands. (d) Both tumours are present, the EMC and the adenocarcinoma, H&E100×. (e) The adenocarcinoma (right) was in close relation with the EMC (left), in an intermingled pattern, H&E 150×. (f) Pancytokeratin Cam5.2 staining the adenocarcinoma. The EMC does not show any staining. H&E 150×. (g) Vimentin stains the EMC, the malignant glands do not show any staining (EMC – extraskeletal myxoid chondrosarcoma).")
(a) The slit corresponded to a diverticulum mainly with ileal type mucosa lining, H&E20×. (b) Some heterotopic gastric type glands were also present (arrowheads) and foci of dysplasia (arrow). The majority of the diverticulum was affected by the extraskeletal myxoid chondrosarcoma (EMC) (star). (c) (Inlet) – heterotopic gastric type glands. (d) Both tumours are present, the EMC and the adenocarcinoma, H&E100×. (e) The adenocarcinoma (right) was in close relation with the EMC (left), in an intermingled pattern, H&E 150×. (f) Pancytokeratin Cam5.2 staining the adenocarcinoma. The EMC does not show any staining. H&E 150×. (g) Vimentin stains the EMC, the malignant glands do not show any staining (EMC – extraskeletal myxoid chondrosarcoma).
An immunohistochemical (IHC) study was performed, evidencing that the myxoid tumour was positive for vimentin (Fig. 3G) and synaptophysin without expressing myogenin, protein S100, PAX8, desmin, chromogranin-A, Melan A, and HMB45. The adenocarcinoma component was positive for Cam5.2 (Fig. 3F) and CK7 without TTF1, synaptophysin, chromogranin-A, and CK20 expression.
The diagnosis of the adenocarcinoma was straightforward based on morphology alone. In the mesenchymal tumour, in the differential diagnosis melanoma, perivascular epithelioid cell tumour (PEComa), epithelioid gastrointestinal stromal tumour, and malignant gastrointestinal neuroectodermal tumour were considered, which were convincingly disproved by the IHC panel. In combination with the past history of EMC, the diagnosis of EMC metastasis to an MD adenocarcinoma was rendered.
The patient was discharged one week after surgery. At that time, she had multiple pulmonary metastases. The patient died eight months later due to disease progression.
DiscussionEMC is a sporadic soft tissue sarcoma, recognized by Sout and Verner in 1953, with the clinical and pathological characteristics defined by Enzinger9 in 1972. Clinically, EMC has a slight male predominance and arises more commonly in the extremities in the fifth and sixth decade of life.2 In our case, age and tumour location were in accordance.
On histological evaluation, EMC typically has a multinodular architecture with fibrous septa and is composed of uniform and interconnected cells, comprising clusters and cords, supported by a myxoid or chondromyxoid matrix. There is low mitotic activity, the stroma is hypovascular, and no discernible cartilaginous histology is observed. High-grade tumours have more undifferentiated solid areas with epithelioid morphology.10 The histological phenotype in our case had increased cellularity and a reduction of the amount of matrix with the lesion progression, but exhibited an overlap with the literature description, translating high-grade dedifferentiation.1,10
Initially, EMC was considered a cartilaginous tumour, but now it is classified as a mesenchymal tumour of uncertain differentiation.1 An interesting and distinctive feature of EMC is the recurrent genetic rearrangements involving the NR4A3 gene on chromosome 9, which may be a valuable confirmatory diagnostic clue. The most frequent fusion partner is EWSR1, but other rare transcripts have been described.11 This genetic alteration has been the basis for the re-designation of this entity as “NR4A3 rearranged myxoid sarcoma” (provisional designation).10 Unfortunately, the genetic study was not performed in our case.
Regarding biological behaviour, EMC is mainly a disease with an indolent behaviour, slow growth, and aggressive potential.10 However, studies with longer follow-ups indicate a high rate of local recurrences and distant metastases.2 When metastases occur, they are more frequent in the lung, followed by bone and lymph nodes.12 Despite this aggressive behaviour, there is a high survival at five years (80%) and ten years (60%) in some cohorts.11 Our case complied with this behaviour, with both local recurrence and distant pulmonary metastases. Despite the fatal outcome of our patient, recurrence occurred five years after the initial diagnosis, which is consistent with the overall survival reported in the literature.
After the leg amputation, the patient received a treatment regimen consisting of doxorubicin, ifosfamide, cisplatin and 49 cycles of trabectedin, after which she had a recurrence in the arm. She underwent 4 cycles of trabectedin and gemcitabine, carboplatin, and etoposide, after which the volvulus was discovered. As a palliative treatment, after the discovery of the lung metastases, the patient was treated with pazopanib and sirolimus.
Regarding treatment for EMC, surgery with negative margins is the standard of care, with a direct impact on the recurrence rate2; nevertheless, even with adjuvant radiotherapy, the relapse rate is 50%.10 In the setting of metastatic disease, there are no factors to guide the clinical decision. Standard anthracycline-based chemotherapy, a common agent for treating advanced soft tissue sarcoma, is of limited action in EMC. Anti-angiogenic drugs and the tyrosine kinase inhibitor (TKI) pazopanib may be promising.13
MD is a congenital anomaly, the most frequent in the gastrointestinal tract.3 A true diverticulum has all the layers of the bowel wall. From a clinical perspective, it has a very interesting rule, the rule of 2's – it affects 2% of the population; symptoms are present in up to 2% of the patients; usually found at 2ft from the ileocecal valve; symptoms typically are more evident after 2 years, heterotopic tissue is present in 50% of cases, and the male/female ratio is 2 to 1.3
Malignancy in the MD is rare, with an incidence of 0.5–3.2%, the prognosis depends mainly on the biological aggressiveness of the neoplasm, and no defined staging classification has been devised.6 Most tumours described in MD are neuroendocrine in origin, followed by mesenchymal tumours, namely GISTs and leiomyosarcomas, and adenocarcinomas.5,13 Some rarer neoplasms, such as lymphomas and melanomas, were also described,5 and secondary metastases to MD reports are present in the literature as case reports.5
Due to the risk of malignization of the MD, a cohort from the Mayo Clinic reported a primary malignancy of 5.1%, and up to one-third developed metastases. Surgical removal upon discovery is recommended.13
In our case, MD adenocarcinoma was an incidental finding during an emergency surgery for a volvulus. No record of metastatic disease of the adenocarcinoma was reported, except for the diffuse metastatic setting of the chondrosarcoma, which dictated the patient's prognosis. The main differential diagnosis for adenocarcinoma would be primitive versus secondary. However, the absence of adenocarcinoma in other locations (radiologic features of the pulmonary lesions were overlapping with the sarcoma features), the dysplasia in the MD, and the immune profile established a primary location in the diverticulum.
Therefore, we describe a unique case of an EMC metastasis to an MD, which coincidently presented adenocarcinoma, and, as far as the authors are aware, has not been reported in the literature.
The pathophysiology could be related to the angiogenic profile of the adenocarcinoma, which could have attracted the mesenchymal tumour, in a phenomenon known as tumour-to-tumour metastasis.
We conducted the literature review according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines and searched PubMed, Google Scholar, and Web of Science, using the following words: Meckel's diverticulum; Meckel's diverticulum adenocarcinoma; metastases; Meckel's diverticulum metastases.
Authors’ contributionsJG and RCO were responsible for data gathering and manuscript writing. RCO was responsible for the diagnosis. MAC, RF and JC were responsible for work supervision, diagnosis and manuscript review. All authors read and approved the manuscript final version for publication.
Ethical considerationsThis study was performed in accordance with the ethical standards defined by the authors’ institution. The samples stored in anatomical pathology departments were collected for diagnostic purposes to promote and safeguard patient health and not as an experimental procedure on patients. Informed consent was obtained for publication.
Patient consent statementVerbal informed consent was obtained from legally authorized representatives before the study.
Financial supportNone.
Conflict of interestThe authors have no relevant financial or non-financial interests to disclose.

