




La alcaptonuria es un trastorno hereditario del metabolismo de la tirosina y la fenilalanina, de transmisión autosómica recesiva, ocasionado por un déficit de la enzima oxidasa del ácido homogentísico (HGO), que provoca su eliminación excesiva por la orina y la acumulación de pigmentos derivados de éste en el tejido conectivo (ocronosis).
La prevalencia es de 1/200.000, en homocigotos. Las manifestaciones clínicas más frecuentes y características son la coloración negroparduzca de la orina al alcalinizarla o al contacto con el aire (por oxidación del ácido homogentísico), la pigmentación ocre de escleróticas, piel y pabellones auriculares y la calcificación de los discos intervertebrales en los estudios de imagen.
El diagnóstico se realiza basándose a los hallazgos clínicos, radiológicos y por la determinación de ácido homogentísico en orina, suero y tejidos.
Presentamos un nuevo caso de ocronosis y artropatía ocrónotica de un paciente varón de 60 años, por su rareza y reconocimiento tardío.
Caso clínico
Paciente varón de 60 años de edad, con historia de aproximadamente 15 años de evolución de raquialgias acompañadas de severa limitación funcional de los 3 segmentos vertebrales y gonalgia bilateral mecánica, con derrames articulares recidivantes.
La evolución fue tórpida, con períodos sintomáticos de duración variable, que mejoraban parcialmente con antiinflamatorios no esteroideos (AINE) y fisioterapia.
En los últimos meses presentaba disnea de pequeños/moderados esfuerzos.
Exploración
A la exploración destacaba: pigmentación ocre de escleróticas (fig. 1), pabellones auriculares, pulpejos de dedos de manos e hiperpigmentación de axilas; disminución global de la movilidad del raquis en sus 3 segmentos Schober, 1 cm; distancia dedo-suelo, 35 cm; expansión torácica, 3 cm, pérdida de la lordosis lumbar fisiológica; limitación dolorosa de la flexoextensión de ambas rodillas y derrame articular en ambas; ACP, soplo pansistólico II-III/VI. Resto de la exploración sin interés.

Figura 1.Pigmentación ocre de escleróticas.
Pruebas complementarias
Laboratorio: hemograma, bioquímica general (20 parámetros) y elemental de orina sin alteraciones, velocidad de sedimentación globular (VSG): 20; proteinograma, balance fosfocálcico y parathormona (PTH) normales, perfil férrico normal; factor reumatoide (FR) y antígeno de histocompatibilidad (HLA) B-27 negativos; líquido sinovial: 150 células, predominio mononucleares, abundantes cristales de pirofosfato cálcico intra y extracelulares; ácido homogentísico en orina de 24 h: 1.540 mg/24 h (referencia < 10 mg/24 h.), prueba de alcalinización de la orina positiva (fig. 2).

Figura 2.Orina normal y alcalinizada.
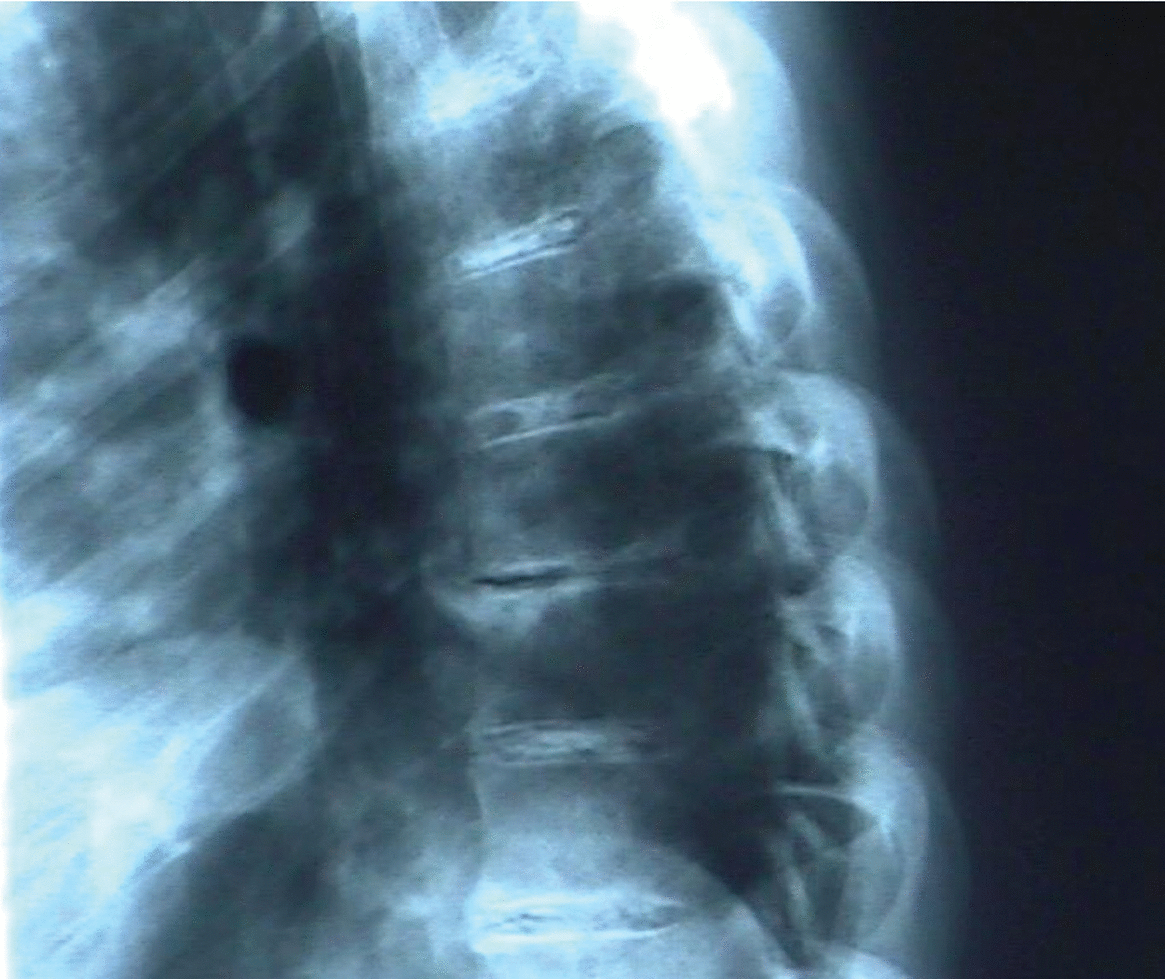
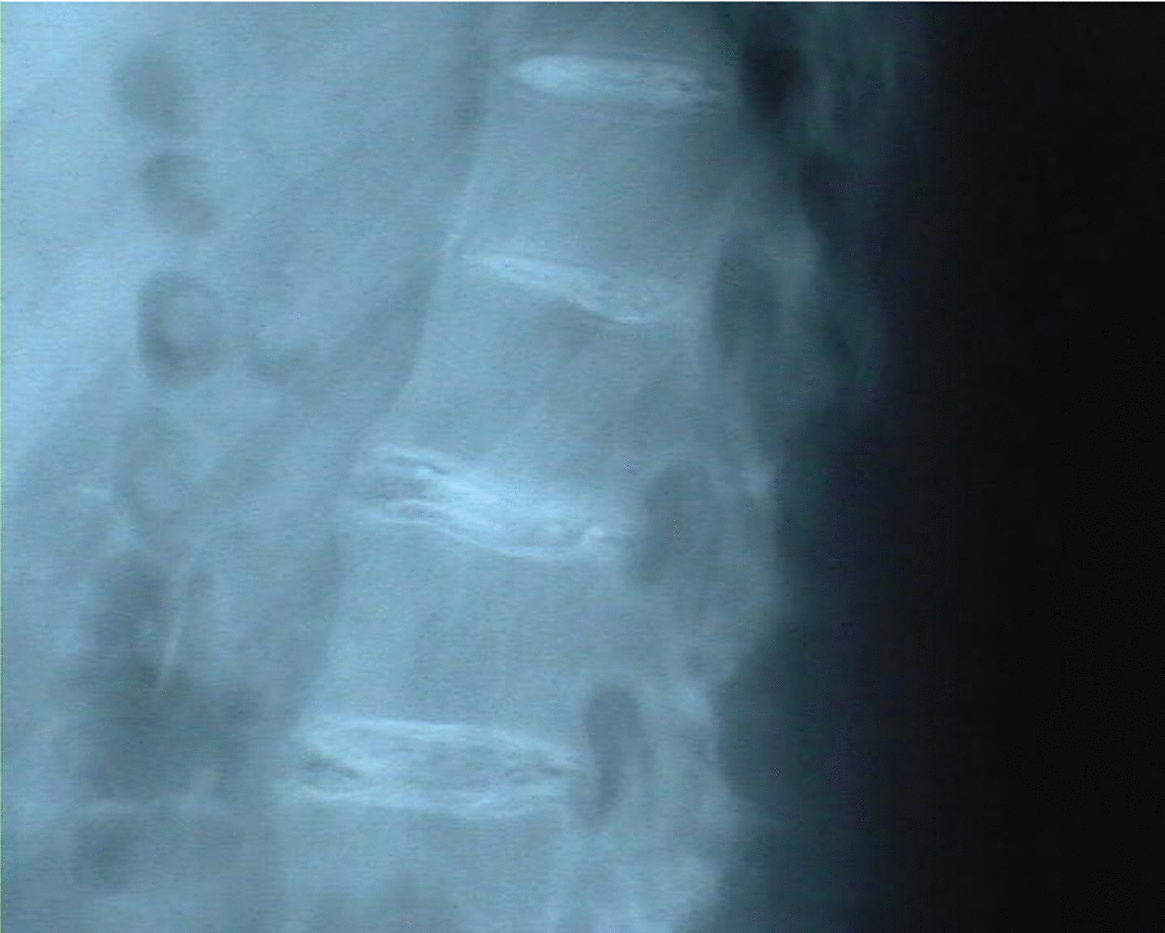
Radiología: calcificaciones discales múltiples en segmentos dorsal y lumbar (figs. 3 y 4); irregularidad de sacroilíacas.
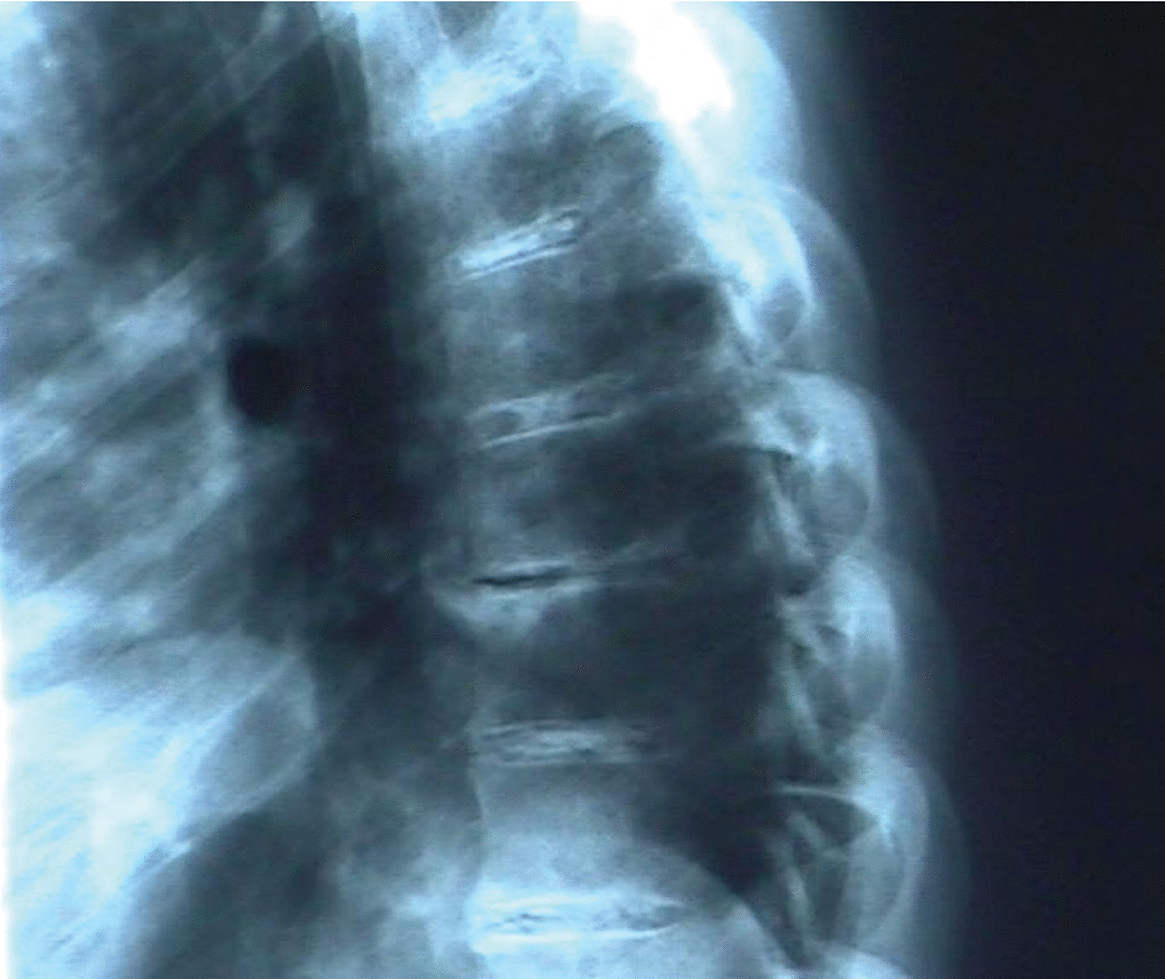
Figura 4.Calcificaciones discales en columna lumbar.
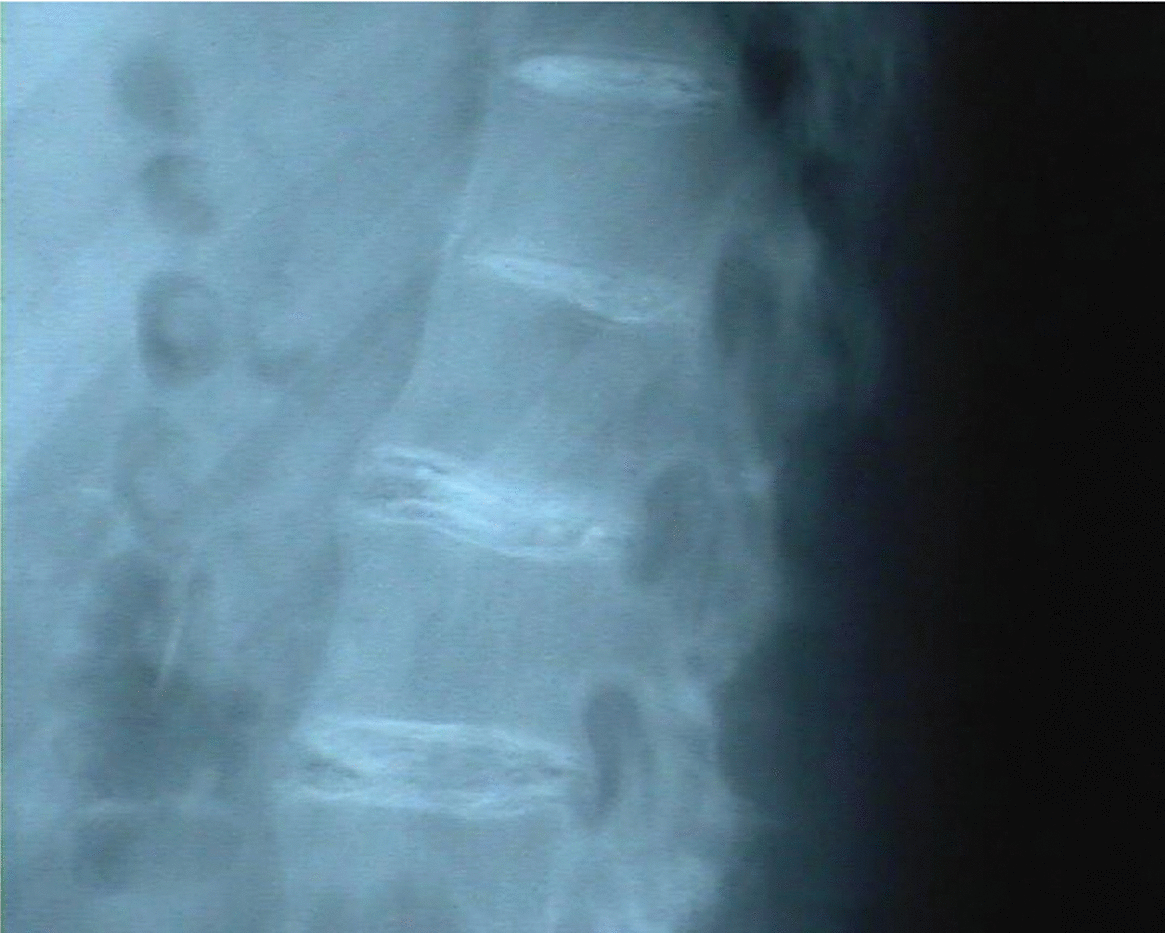
Figura 3.Calcificaciones discales en columna dorsal.
Otras pruebas: histología de biopsia cutánea: haces colágenos degenerados y homogeneizados, teñidos de color ocre (fig. 5); ecocardiograma, calcificación mitral y estenosis aórtica; sometido a reemplazo valvular, se objetivan depósitos ocres en válvulas protetizadas.
Discusión
La alcaptonuria es un trastorno hereditario del metabolismo de la tirosina y fenilalanina. Fue la primera enfermedad humana en la que se demostró una herencia autosómica-recesiva.
Se caracteriza por un déficit de la enzima oxidasa del ácido homogentísico (HGO)1, encargada de trans-
formarlo en ácido maleilacetoacético, que como consecuencia ocasiona su eliminación excesiva por la orina y la acumulación de los pigmentos derivados de éste en el tejido conectivo (piel, escleróticas, pabellones auriculares, traquea, válvulas cardíacas y cartílago articular), dando lugar a la ocronosis2.
Suele presentarse en el 50% de los pacientes con alcaptonuria y siempre a partir de la cuarta década de la vida3.
Hasta 16 tipos de mutaciones en el gen de HGO humano pueden dar lugar a una pérdida de función de la enzima4.
La prevalencia es de 1/200.000, en homocigotos. Los portadores heterocigotos no presentan la enfermedad.
Se ha observado una posible asociación de la alcaptonuria con el HLA B-275.
Dentro del amplio espectro de manifestaciones clínicas destacan, como características, inicialmente la presencia de orinas oscuras o negroparduzcas, que se originan por la oxidación del ácido homogentísico de la orina al contacto con el aire o medio ambiente y que habitualmente no objetiva el paciente, al no precisar de su almacenamiento.
Esto origina el retraso en el diagnóstico. Posteriormente, y pasados los años, aparecen los síntomas y signos patognomónicos de la ocronosis, consecuencia del depósito de los pigmentos del ácido homogentísico en tejido conectivo, que originan una coloración ocre (de ahí su nombre) de las escleróticas (fig. 1), pabellones auriculares, oído interno, piel, válvulas cardíacas, traquea y cartílagos articulares6,7.
La afectación de cartílagos articulares, discos, etc., va a originar la artropatía ocronótica, en sus formas axial y periférica2.
La afectación axial, fundamentalmente dorsal y lumbar, tambien en cervical, se presenta como dolor de tipo mecánico, acompañado de limitación funcional (espondilosis ocronótica), con la presencia en la radiología de las características calcifi-
caciones discales múltiples2,8, la afectación de interapofisarias y de sacroilíacas (pinzamiento y esclerosis), es más rara2.
En las articulaciones periféricas, sobre todo rodillas, caderas y hombros, se presenta como cuadro de dolor, también de tipo mecánico, por proceso degenerativo del cartílago articular, que puede acompañarse de derrames articulares no inflamatorios. Se puede observar la presencia de cristales de pirofosfato y apatita en el líquido sinovial6,7.
La presencia de calcificaciones y estenosis valvulares cardíacas, por depósito de material ocronótico, es muy frecuente9-11, con reemplazo valvular en algunos casos12, como fue el caso del paciente que aquí presentamos.
Se han descrito cálculos prostáticos de color negruzco13.
El diagnóstico se establece basándose en los hallazgos clinicoradiológicos previamente comentados y la determinación de ácido homogentísico en orina y plasma por métodos enzimáticos y colorimétricos específicos14.