




Introducción
La artrosis, enfermedad articular degenerativa u osteoartritis en terminología anglosajona (OA), es el resultado de un desequilibrio en la homeostasis condrocitaria. Como resultado, se produce un desequilibrio o falta de acoplamiento entre degradación y síntesis de los distintos componentes de la matriz extracelular, con un claro predominio de los procesos catabólicos. Aunque se considera al condrocito como el elemento clave en el mantenimiento de dicha homeostasis, otras estructuras participan también en el inicio y progresión de la enfermedad. De hecho, hoy se habla de OA como de fracaso o insuficiencia global de la articulación, donde participan todas las estructuras articulares, desde el cartílago hasta la cápsula y los ligamentos, teniendo especial importancia la interacción entre estos 3 elementos: cartílago hialino normal, hueso subcondral y membrana sinovial, esta última especialmente en las fases más evolucionadas de la enfermedad1-3. En este artículo, nos centraremos precisamente en el papel que tanto la sinovial como el hueso subcondral juegan en la patogenia de la OA, así como sus posibles implicaciones terapéuticas.
Hueso subcondral
El hueso subcondral (HS) comprende el tejido subarticular mineralizado que se extiende desde el tidemark (frente de mineralización o unión entre el cartílago calcificado y no calcificado) hasta el inicio de la médula ósea. Sus funciones principales consisten en dar soporte al cartílago articular suprayacente, distribuir la carga mecánica a la diáfisis cortical subyacente, así como absorber la tensión de los impactos mecánicos continuos y nutrir las capas profundas del cartílago hialino, especialmente en el período de crecimiento4.
El HS incluye al menos 3 estructuras mineralizadas bien diferenciadas: el cartílago calcificado, un hueso laminar subcondral corticalizado y el hueso subcondral trabecular. El cartílago calcificado junto con el hueso cortical subcondral son conocidos en la bibliografía como placa subcondral4,5. Algunos autores incluyen también un hueso trabecular subarticular cuyos límites no están bien definidos. El espesor del HS varía en función de la especie animal, edad, masa corporal, localización y tipo de articulación. Así, el grosor del HS en el platillo tibial humano puede llegar hasta 2-3 mm en la zona de más carga. El HS está muy vascularizado, aunque la mayoría de los vasos no alcanzan el cartílago calcificado, y excepto en la enfermedad, ninguno penetra en el cartílago hialino4. El cartílago calcificado puede aumentar de espesor mediante la osificación endocondral, contribuyendo a la esclerosis subcondral observada en las radiografías. Asimismo, el HS puede incrementar el grosor por aposición directa de hueso (modelado) y aumentar su densidad a través del remodelado óseo, factores ambos que incrementan la densidad aparente de dicha esclerosis subcondral. En condiciones normales, el crecimiento, modelado y remodelado óseos ocurren constantemente a lo largo de la vida, pero su actividad varía en diferentes grados en los 3 tejidos mineralizados, con diferente resultado sobre la masa y geometría del hueso y diferentes consecuencias en la mecánica articular5. El contenido mineral y características densitométricas del HS no
están bien definidos en la bibliografía. Nosotros hemos determinado el contenido y densidad mineral ósea (DMO) en esta región en un modelo animal, encontrando valores de DMO intermedios entre el hueso trabecular y el cortical6.
El proceso de osificación endocondral ocurre a lo largo de la vida, dando lugar en la OA a un avance del tidemark con pérdida de su perfil ondulado, duplicidad del frente de mineralización y engrosamiento del cartílago calcificado hasta en un 25% del espesor total del cartílago (en condiciones normales la relación entre ambos suele ser de 10 a 1)7. Estos cambios confieren mayor rigidez al cartílago calcificado, lo que conlleva una mayor sobrecarga del cartílago hialino suprayacente, favoreciendo de esta forma la iniciación y progresión de la OA.
Llegado este punto, conviene matizar que existe una distinción entre densidad ósea aparente, definida como masa ósea/volumen total, y densidad ósea mineral, definida como masa ósea/volumen óseo. Li y Aspden8 muestran que, aunque la densidad aparente del HS es significativamente mayor en los pacientes artrósicos que en los individuos normales u osteoporóticos, la densidad mineral es significativamente menor debido a una hipomineralización de dicho tejido. Es esta densidad aparente la que normalmente vemos en las radiografías de los pacientes con artrosis y esclerosis subcondral.
Esto hace plantearnos una cuestión debatida y no resuelta en la literatura acerca de la posible relación existente entre 2 entidades muy prevalentes: artrosis y osteoporosis, y que tendría importantes y evidentes implicaciones terapéuticas.
Hoy día, y aunque existen opiniones contrapuestas, la mayoría de estudios clínicos sugieren una relación inversa entre osteoporosis y OA9-11. En el estudio Framingham, la DMO de cadera fue el 5-9% mayor en mujeres con OA grado 1-2 que en controles sin OA12. Esta relación inversa entre artrosis y osteoporosis podría reflejar una diferencia cualitativa en cuanto a la calidad ósea entre ambas. No obstante, existen algunas variables de confusión que podrían explicar per se esta misma relación mutuamente excluyente, como la raza, el sobrepeso y la actividad física13. Así, las personas obesas o con excesiva actividad física tendrían mayor riesgo de artrosis a la vez que un incremento de la masa ósea. Existen, además, determinados sesgos de selección en estos estudios clínicos que invalidan o crean cierta incertidumbre respecto a los resultados obtenidos. Así, la mayoría de estos estudios no son aleatorios, en algunos incluso faltan controles pa-reados apropiados, los tamaños muestrales fueron a veces extremadamente pequeños y el diagnóstico de OA se basaba exclusivamente en la clínica, sin confirmación radiológica en algunos de ellos4.
Radin y Rose14 ya propusieron, hace años, que el incremento en la densidad del HS aumentaría paralelamente la rigidez de dicho tejido. Esto produciría una disminución o pérdida de sus propiedades viscoelásticas, reduciendo su capacidad de amortiguación con la consiguiente sobrecarga del cartílago suprayacente, e iniciándose así un deterioro progresivo del cartílago, como ya hemos comentado15. Es la base de la teoría biomecánica. Posteriormente y según progresa la artrosis, se iría produciendo una desmineralización y osteoporosis multifactorial que ocasionarían la aparición de microfracturas en el HS y microcracks en el cartílago calcificado, cuyo intento reparador potenciaría la angiogénesis con formación e invasión de nuevos vasos que
irían penetrando en el cartílago calcificado con aumento de su espesor, esclerosis subcondral y progresión de la OA4,5. Esto daría lugar a un hueso y cartílago aún más rígidos, con perpetuación del proceso degenerativo. Este crecimiento vascular iría asociado con la producción de distintos factores angiogénicos que a su vez activarían metaloproteasas (MMP) inactivas que ocasionarán mayor resorción del cartílago y calcificación de la placa subcondral.
De una forma resumida, de los estudios existentes, podríamos concluir que la existencia de osteoporosis podría prevenir o retardar el inicio de la artrosis, pero posteriormente haría empeorar y progresar la enfermedad de manera más evidente.
Recientemente, el grupo de Pelletier16 ha sugerido una hipótesis interesante que postula un incremento en el remodelado óseo del HS como base patogénica de la artrosis16. Siguiendo el esquema propuesto por estos autores, un aumento del remodelado óseo subcondral conduciría a un deterioro o pérdida de la matriz extracelular cartilaginosa e iniciaría un intento de reparación estructural por parte tanto del propio cartílago (condrocitos) como del HS subyacente. En este punto crítico, puede ocurrir que los procesos anabólicos y restauradores controlen/reparen el daño establecido, o se vean sobrepasados, dando lugar al desarrollo y progresión de la OA (fig. 1).
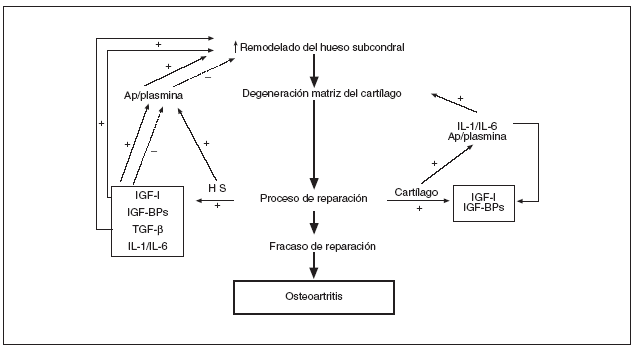
Figura 1. Representación esquemática de los mecanismos involucrados en el remodelado del hueso subcondral OA y posible interacción con el cartílago (reproducida de cita 16). Ap/plasmina: activador del plasminógeno-plasmina; IGF-I: factor de crecimiento insulínico 1; IGF-BP: proteínas de unión al IGF; TGF-ß: factor transformador del crecimiento ß; IL-1 y 6: interleucinas 1 y 6; HS: hueso subcondral.
La confirmación de esta secuencia de hechos podría tener importantes implicaciones terapéuticas, en un intento de frenar dicho remodelado con fármacos antirresortivos. De hecho, existen estudios observacionales que demuestran la utilidad de los estrógenos para ralentizar la progresión de la OA tras la menopausia, aunque más interés tendrían los bisfosfonatos y algunos antiinflamatorios no esteroideos con capacidad de inhibir la resorción ósea, como luego comentaremos.
Papel de la membrana sinovial en la artrosis
Hasta hace poco se consideraba la afectación del tejido sinovial en la artrosis como mínima y secundaria al daño originado en el cartílago articular. Ahora se sabe que en las fases finales de la OA, la membrana sinovial desarrolla una respuesta inflamatoria que contribuye de manera decisiva en la patogenia y grado de expresividad clínica de la enfermedad3,17. Los cambios morfológicos observados en la sinovial artrósica en las fases avanzadas de la enfermedad muestran proliferación de células mononucleares, incluyendo linfocitos T y en menor grado B activados, hiperplasia sinovial, aumento de la angiogénesis, junto a un incremento de la osteoclastogénesis en las zonas de erosión ósea, muy similares a los encontrados en las artropatías inflamatorias y, en particular, en la artritis reumatoide (AR). También se han descrito diversos grados de sinovitis en los estadios tempranos de la OA18. De hecho, la presencia de inflamación sinovial en la OA puede ser de suma importancia en el proceso de cronicidad y autoperpetuación en el deterioro del cartílago articular, dado que la sinovial activada sintetiza y libera al medio múltiples mediadores de inflamación, como proteasas, citocinas catabólicas proinflamatorias, mediadores lipídicos y radicales libres, que intervienen y modulan el metabolismo condrocitario, con un claro predominio de los fenómenos catabólicos3,17 (tabla 1). Entre todos los mediadores de inflamación que intervienen en el catabolismo del cartílago articular, cabe destacar el papel de algunas MMP (colagenasas 1 y 3, estromelisinas, etc.) y agrecanasas, citocinas catabólicas de la familia de la IL-1 (IL-1*, IL-1ß y IL-1Ra) y el factor de necrosis tumoral (TNF), radicales libres como el óxido nítrico (ON) y eicosanoides como PGE2 y LTB4. Posteriormente, se origina un círculo vicioso cerrado que conduce a un mayor deterioro del cartílago, liberación de más mediadores proinflamatorios y destrucción progresiva de la matriz extracelular. En las fases avanzadas de la artrosis, se liberan pequeños fragmentos de colágeno a la cavidad articular, así como microcristales provenientes de la degradación del cartílago, y los que se encuentran más a menudo son los de pirofosfato cálcico y los de hidroxiapatita19. Todos estos fenómenos inducen la síntesis de más citocinas proinflamatorias por parte de fibroblastos y macrófagos, que perpetuarán y abocarán a un proceso irreversible de degeneración del cartílago articular. Por otra parte, la membrana sinovial OA aumentaría también la síntetis de citocinas reguladoras y antiinflamatorias como IL-4, IL-10 e IL-13. Estas citocinas disminuirían la producción de IL-1ß, TNF-* y MMP, inhibirían PGE2, e incremetarían los valores de TIMP-1 y de IL-1Ra20,21. El tejido sinovial es capaz también de sintetizar IL-6, importante factor responsable de la destrucción ósea.

Hueso subcondral y membrana sinovial como diana terapéutica en la artrosis
Si tenemos en cuenta la hipótesis expuesta con anterioridad respecto al incremento del remodelado óseo como factor patogénico previo al deterioro inicial del cartílago, parece razonable pensar que los fármacos antirresortivos óseos podrían ser de utilidad. Sin embargo, si la rigidez ósea subcondral fuera el factor patogénico predominante para el desarrollo de OA, probablemente estos fármacos se-rían perjudiciales, en tanto que aumentan la masa ósea al disminuir el remodelado. Como efecto adicional, y dado que tanto el modelado como el remodelado óseos son responsables de la adaptación geométrica de la articulación a las nuevas condiciones laborales, estos agentes antirresortivos podrían evitar esta nueva adaptación articular con el consecuente efecto deletéreo sobreañadido. No obstante, y a pesar de las consecuencias teóricas adversas, lo cierto es que existen estudios en diversos modelos animales y en humanos que apoyan justo lo contrario, apuntando a los antirresortivos como alternativa de utilidad para el tratamiento de la OA.
Así, el ácido zoledrónico se ha mostrado eficaz para prevenir el daño cartilaginoso en un modelo de OA de rodilla inducida por quimiopapaína en conejos22. Asimismo, se ha visto que el risedronato puede conservar las propiedades biomecánicas ligamentarias en un modelo de OA de rodilla inducida por resección del ligamento cruzado anterior en conejos (XLCA)23; pero el estudio más consistente es el de Hayami et al24, que demuestra la utilidad del alendronato para prevenir la osteofitosis, evitando la progresión del daño cartilaginoso en ratas. En humanos, el etidronato ha mostrado también un efecto analgésico beneficioso en espondilo y gonartrosis, y el clodronato administrado en inyección intraarticular ha demostrado eficacia para reducir la sinovitis secundaria a OA avanzada al disminuir los valores de PGE2 dentro de la articulación25,26.
Otro grupo de fármacos que ha mostrado cierta utilidad como antirresortivos y «condroprotectores» son algunos antiinflamatorios no esteroideos (AINE) como carprofen y los inhibidores duales del sistema COX/LOX. En concreto, y en estudios preliminares, carprofen parece modificar la progresión de la OA experimental27. En un estudio de OA inducida en perros, carprofen reducía el deterioro del cartílago a lo largo del tiempo, a la vez que disminuía la resorción ósea y el número de unidades de remodelado de forma significativa. A nivel bioquímico, se apreciaba una reducción en los valores de IGF-1, factor activador del plasminógeno y una disminución en la producción de TGF-ß por los osteoblastos de los animales tratados con carprofen27.
Hoy día se están desarrollando nuevos AINE, entre los que destacan los inhibidores de leucotrienos y los inhibidores duales del sistema COX/LOX, entre los que licofelona ocupa un lugar destacado28. En el HS de modelos caninos de artrosis provocada por sección del LCA, licofelona ha mostrado un claro efecto antirresortivo, reduciendo además los valores de metaloproteasa 13 y catepsina K, lo que podría interpretarse como «efecto condroprotector antiartrósico», aunque harían falta más estudios que confirmen estos datos preliminares29.
Teniendo en cuenta los hallazgos comentados en la membrana sinovial, no vamos a insistir en la importancia de ésta como diana terapéutica en la artrosis. En la sinovial, los hallazgos de un gran número de estudios indican que el objetivo más interesante e inmediato sería bloquear el efecto de las citocinas catabólicas más relevantes en la patogenia de la OA e incrementar o potenciar los antagonistas de los receptores de dichas citocinas.
A continuación, detallaremos algunas de las estrategias más prometedoras en el tratamiento de la OA en la sinovial (tabla 2):

Inflamación y citocinas. Dado que la IL-1ß, amén de otras citocinas y mediadores inflamatorios (TNF, óxido nítrico, etc.), tiene un papel patogénico relevante o central en la progresión de la OA, parece razonable utilizar agentes que inactiven su actividad biológica30-32. Así, uno puede reducir su actividad celular mediante la utilización de antagonistas del receptor de IL-1 o mediante el empleo de sus receptores solubles33. Las acciones de esta familia de citocinas pueden también reducirse bloqueando algunos de los mecanismos de señalización intracelular34,35. Varios estudios in vitro han demostrado que IL-1Ra es capaz de reducir varios de los procesos catabólicos dependientes de IL-1. Asimismo, la inyección intraarticular de IL-1Ra parece reducir la progresión de la OA en modelos animales36. Recientemente, existen datos que muestran que la transfección del gen de IL-1Ra a rodillas artrósicas de perros y conejos, reduce la progresión de las lesiones artrósicas37,38. Otra interesante diana para inactivar el sistema de IL-1 es mediante la inhibición de la ECI, enzima de conversión de IL-1ß o caspasa-1. Como es sabido, la ECI transforma la proforma inactiva de IL-1 en su forma activa madura. Esto aportaría una base racional para la utilización de inhibidores de la ECI en el tratamiento de la OA39. Hoy día existen estudios que emplean este tipo de inhibidores por vía oral en pacientes con AR, con resultados esperanzadores. Existen también estudios que prueban la utilización de los distintos receptores solubles de IL-1 y TNF-*. Otra alternativa terapéutica sería la utilización de citocinas reguladoras antiinflamatorias, como IL-4, IL-10 e IL-13.
Mecanismos de señalización intracelular. Otra forma de actuación para contrarrestar los efectos catabólicos de las citocinas proinflamatorias incluye la inhibición de los mecanismos de señalización intracelular en la cascada de las proteincinasas o de los factores de transcripción nuclear. Así, el inhibidor selectivo de P38 ha mostrado reducir la progresión estructural en un modelo de artritis inflamatoria en ratas40. Del mismo modo, la inhibición selectiva de ERK 1/2 por vía oral podría reducir la progresión de OA en un modelo experimental en conejos, al disminuir la síntesis de MMP41. Asimismo, existen interesantes estudios que intentan bloquear la unión nuclear de los factores de transcripción como NF-* B y AP-142.
Metaloproteasas. La inhibición de las MMP podría considerarse el punto central en el tratamiento de la OA, incluso hasta poder considerar como verdaderos fármacos modificadores de OA a los fármacos que actúan a este nivel. En este sentido, habría que incluir determinados inhibidores químicos, como los quelantes de metales pesados v.gr.: cinc y calcio, o las tetraciclinas (mino y doxiciclina). Sin embargo, este modo de actuación no está exento de frecuentes efectos adversos, que obligan a investigar inhibidores selectivos de MMP específicas.
Radicales libres y COX-2. El ON y sus subproductos son capaces de inducir no sólo inflamación local, sino también daño y destrucción tisular a través de múltiples mecanismos43. En primer lugar, el ON induce muerte condrocitaria por apoptosis44. También pueden estimular la síntesis y actividad de diversas MMP e inhibe la síntesis de colágeno y proteoglicanos32,33. El ON es, además, capaz de incrementar la actividad de la COX-2 y secundariamente la respuesta inflamatoria45. Inhibir la producción y exceso del ON se ha convertido, por tanto, en uno de los principales objetivos en el tratamiento sintomático y modificador de la OA. Un estudio reciente muestra la utilidad como modificador de enfermedad de un inhibidor específico de la ON sintetasa administrado oralmente en un modelo de artrosis en perros43. Asimismo, la inhibición selectiva de COX-2 bloquea la apoptosis inducida por ON46. Ya hemos comentado también la utilidad de los inhibidores de los leucotrienos para frenar la progresión de la enfermedad OA.
Como conclusión, podríamos decir que la OA constituye un apasionante campo en expansión para la investigación y el desarrollo de nuevos fármacos. El hueso subcondral y la sinovial representan 2 estructuras fundamentales en la patogenia de la enfermedad, por lo que también constituyen interesantes dianas a la hora de explorar y diseñar nuevos agentes eficaces como modificadores de esta enfermedad.