



Introducción
La artrosis (OA) es una patología degenerativa de las articulaciones que se caracteriza por la degradación del cartílago articular hialino. El envejecimiento del organismo se asocia de forma importante con la presencia de esta patología. Una característica importante de la OA es su lenta progresión, de forma que la pérdida de la integridad articular sólo puede detectarse al cabo de años de evolución. En su fase final, la OA, refleja una insuficiencia de los procesos de reparación del cartílago, y da como resultado la degradación de la matriz extracelular (MEC), muerte de los condrocitos y la pérdida total de la integridad del cartílago1. Sin embargo, el desarrollo de esta patología no sólo afecta al cartílago, sino a toda la estructura articular, incluyendo el hueso subcondral, el tejido sinovial, la cápsula articular y los tejidos blandos periarticulares2 (fig. 1).
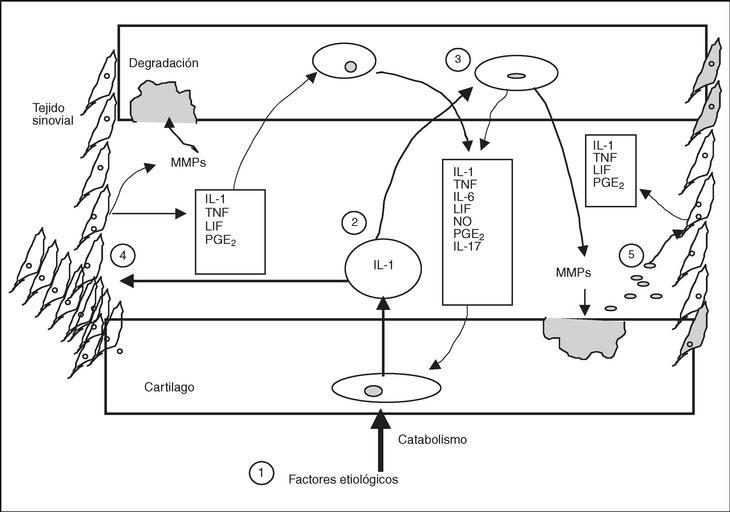
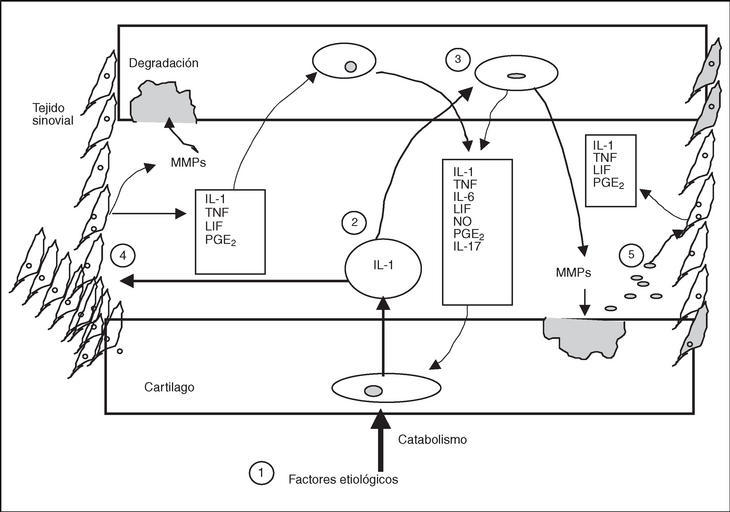
Figura 1. Mecanismos de regulación en el cartílago articular y en el tejido sinovial de la articulación artrósica (únicamente se reflejan los factores catabólicos, de forma simultánea se disparan factores anabólicos compensadores, en la artrosis se produce una prevalencia de los catabólicos: 1) los factores etiológicos, independientemente de cuáles son, desencadenan en el condrocito el programa catabólico; 2) como resultado del programa catabólico existe un aumento de la síntesis de los mediadores proinflamatorios (aquí representamos únicamente a la interleucina [IL] 1 y el factor de necrosis tumoral [TNF]); 3) la IL-1 puede actuar sobre otros condrocitos del cartílago favoreciendo la síntesis de otros factores catabólicos como otras citocinas, el óxido nítrico (NO), las prostaglandinas (Pg) y las metaloproteasas (MMP) que degradan el cartílago; 4) la IL-1 también puede actuar sobre el tejido sinovial, que es capaz de sintetizar MMP que contribuyen a la destrucción del cartílago articular y factores proinflamatorios como otras citocinas y PGE2, que pueden actuar sobre el cartílago y perpetuar el programa catabólico en los condrocitos, y 5) los fragmentos de cartílago liberados al espacio articular como consecuencia de la degradación tienen capacidad para estimular la síntesis de factores inflamatorios por el tejido sinovial. LIF: leukemia inhibitory factor.
La etiología exacta de la OA está lejos de ser totalmente entendida, a pesar de ser la patología reumatológica más común y, probablemente también la patología médica de cuya presencia hay constancia desde hace más años2. Sí, sabemos que los cambios estructurales que observamos en la OA son debidos a la combinación de diversos factores que van desde factores mecánicos a bioquímicos3. Entre los distintos factores que se han identificado últimamente, cabe destacar los factores mecánicos, por ser realmente importantes en el inicio y la evolución de la enfermedad4 y los factores endógenos, como la mutación del colágeno tipo II5.
Clásicamente, el concepto general de OA otorgaba mayor énfasis a la activación directa del cartílago y del hueso subcondral y daba menor importancia al tejido sinovial. Hoy día, sabemos que en las fases finales de la artrosis, la membrana sinovial desarrolla una respuesta inflamatoria que contribuye de manera decisiva en la patogenia y en el grado de expresividad clínica de la enfermedad. En este sentido, los cambios patológicos que se producen en la membrana sinovial de un paciente con un grado severo de OA son próximos a los cambios observados en la membrana sinovial de un paciente con artritis reumatoide (AR)6.
Existen varias teorías que intentan explicar por qué se produce este fallo articular. La teoría más generalizada defiende que es en el cartílago articular donde se produce la pérdida del equilibrio entre el programa catabólico y anabólico del condrocito, lo cual origina el desequilibrio entre la síntesis y degradación de la matriz extracelular del cartílago articular. El resultado final es una destrucción acelerada de la MEC, principalmente por las enzimas proteolíticas procedentes de los propios condrocitos y de las células sinoviales, seguida por alteraciones en los sistemas de reparación del cartílago.
Por último, recientemente ha surgido una nueva hipótesis que, de confirmarse supondría, un nuevo abordaje en el tratamiento de la OA. Esta hipótesis sugiere, que el origen de la OA es consecuencia de un desorden sistémico que afectaría a la diferenciación de las células estromáticas y al metabolismo lipídico. Se basa en una serie de observaciones: la estrecha relación de esta patología con la obesidad, el origen común mesenquimático de las células que constituyen todos los tejidos que forman la cavidad articular, y el posible papel de los mediadores neuroendocrinos (como la leptina) en la regulación de la masa ósea7.
En este capítulo haremos una revisión de aquellos elementos y mecanismos que de alguna forma están involucrados con la OA, haciendo especial énfasis en la OA humana.
Fisiopatología del cartílago articular hialino en la artrosis
Cambios en el número de células y patología artrósica
El condrocito es el único elemento celular presente en el cartílago articular normal, y por tanto desempeña un papel fundamental en el mantenimiento de la integridad de la matriz extracelular del cartílago; así como en la reparación del tejido dañado. La OA se caracteriza por un dramático cambio en el número de células1 (fig. 2). El número de células va a depender del equilibrio entre nacimiento (mitosis) y muerte celular. En los últimos años ha cobrado especial relevancia el papel que la muerte celular pueda desempeñar en la homeostasis celular del cartílago. En este sentido, existen 2 formas de muerte celular: apoptosis y necrosis. La principal diferencia entre ellas radica en que la muerte celular por apoptosis (suicidio celular), no desencadena respuesta inflamatoria. Es un proceso activo bajo control molecular y por ello requiere un consumo de energía. Energía que se emplea en desmantelar de forma ordenada las estructuras celulares impidiendo, de este modo, el daño tisular8.
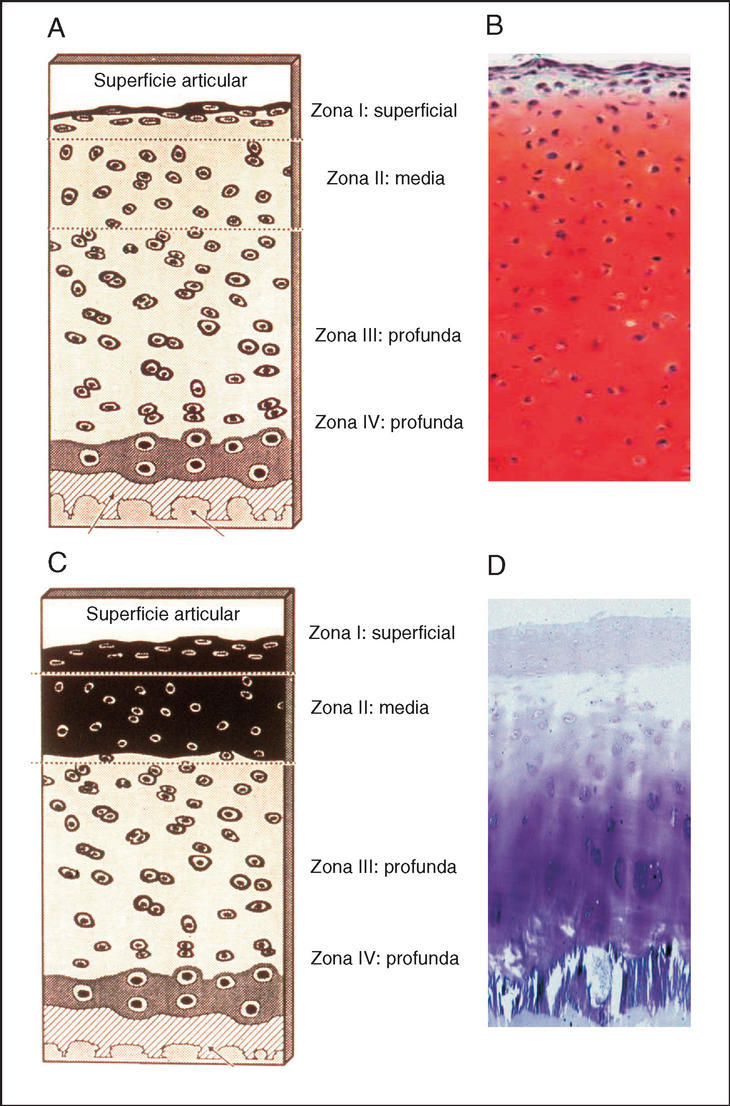
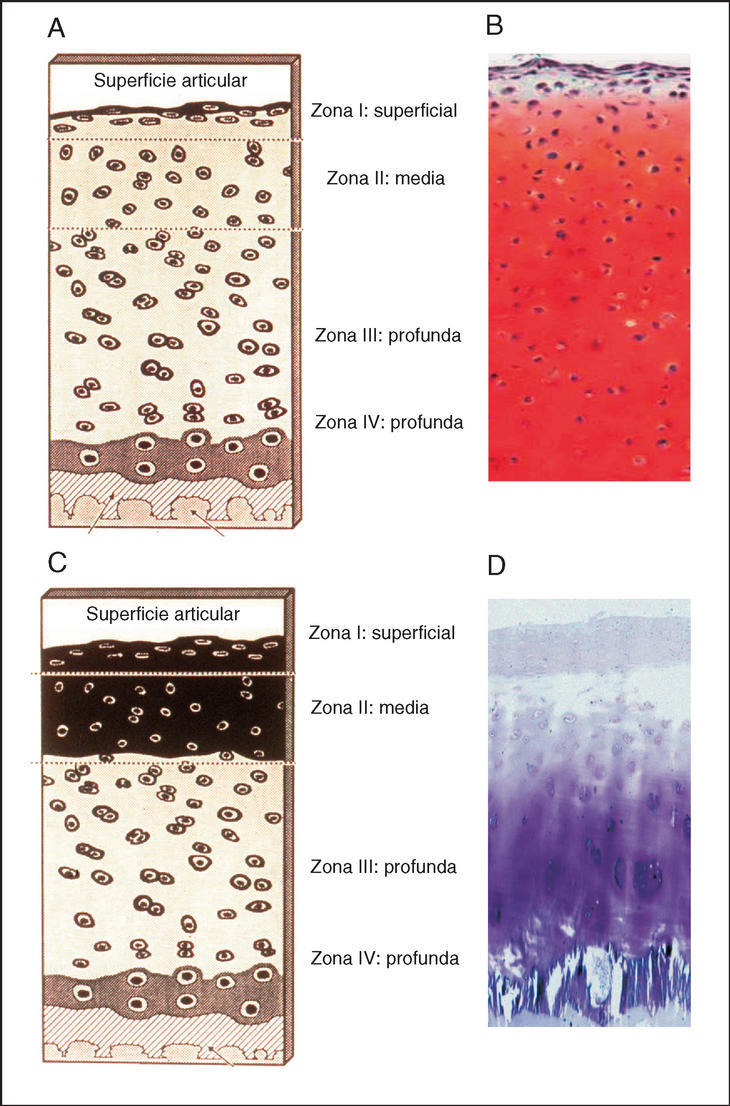
Figura 2. Histología del cartílago articular normal y artrósico. Paneles A y B: en la capa superficial o zona I, los condrocitos son algo más pequeños y aplanados, están situados de forma paralela a la superficie articular. En la zona media o zona II, que representa entre el 60-40% de todo el cartílago, los condrocitos son algo más grandes, esféricos y se encuentran solos o en grupos. En la zona III o profunda, los condrocitos son más elípticos, forman columnas y se orientan perpendicularmente a la superficie. En la zona IV o calcificada, los condrocitos están más esparcidos. Los paneles C y D muestran los cambios típicos del cartílago artrósico, se produce un descenso en el contenido de la matriz extracelular y del número de condrocitos, principalmente en las capas I y II. Esta hipocelularidad se ocasiona por un incremento en la apoptosis de los condrocitos artrósicos.
La muerte celular del condrocito por apoptosis conduce a un cartílago hipocelular que puede ser consecuencia de un programa intrínseco que se activa en los condrocitos senescentes, o que puede ser causa de factores extrínsecos, tales como citocinas que estimulan la degradación de la matriz y/o activan los receptores de muerte celular9. Nuestro grupo ha publicado que los condrocitos procedentes de cartílagos artrósicos tienen las características morfológicas de la apoptosis1. Sin embargo, los mecanismos extracelulares e intracelulares que intervienen en la apoptosis de los condrocitos articulares humanos no son completamente conocidos.
El papel que el óxido nítrico (NO) desempeña como mediador de la patología articular es evidente. En este sentido, se ha demostrado que el NO inhibe la proliferación de los condrocitos e induce apoptosis en los condrocitos articulares humanos10. Cada vez son más numerosos los datos que demuestran el papel clave que desempeña la mitocondria en el desarrollo del proceso apoptótico de los condrocitos11-13. Los resultados obtenidos demuestran que, tanto el exceso de NO que se encuentra en el tejido artrósico, como las alteraciones en la cadena respiratoria mitocondrial que presentan los condrocitos artrósicos, podrían intervenir en la apoptosis del condrocito14.
Durante la apoptosis se activan unas enzimas proteolíticas intracelulares denominadas caspasas. Las caspasas constituyen una familia de cisteína proteasas, que se expresan constitutivamente como zimógenos (procaspasas) y que requieren ser escindidas para ser activadas. Basándonos en su función, las caspasas se dividen en 3 categorías funcionales: caspasas involucradas en la maduración de citocinas (caspasas 1, 4, y 5), caspasas efectoras (caspasas 3, 6, y 7), y caspasas iniciadoras (caspasas 8, 9, y 10)15. La activación de la cascada de las caspasas parece desempeñar un papel esencial en el mecanismo por el cual el NO media la apoptosis en los condrocitos. En esta línea, nuestro grupo ha analizado como el NO, junto con la interleucina-1β (IL-1β) y el factor de necrosis tumoral-alfa (TNF-α), inducen la expresión y la modulación de los diferentes miembros de la familia de las caspasas en condrocitos normales y también en condrocitos artrósicos. El NO, el TNF-α, y la IL-1β inducen la expresión de las caspasas 3, 8, y 7. La caspasa 1 únicamente es inducida por el TNF-α16.
Otra familia de proteínas importantes en relación con la apoptosis es la familia de proteínas de muerte celular Bcl-2. Esta última comprende 2 subfamilias: proteínas antiapoptóticas (subfamilia Bcl-2) y proteínas proapoptóticas (subfamilia bax). En esta línea, diversos estudios demuestran como Bcl-2 en OA desarrolla un papel fundamental en la supervivencia de los condrocitos17.
Finalmente, el ligando de Fas (LFas) es una de las principales citocinas que regulan la apoptosis en diversos sistemas biológicos. En este sentido, recientemente se ha visto que Fas y LFas se expresan en condrocitos articulares, lo que sugiere que la activación de este sistema puede contribuir a la apoptosis en el condrocito. Los condrocitos de la región superficial del cartílago en la OA expresan Fas, que tras la unión de LFas puede inducir la apoptosis del condrocito9. Es de destacar que es precisamente aquí en donde se localizan un mayor número de células apoptóticas. Sin embargo, la concentración de LFas en líquido sinovial artrósico es muy baja.
Mediadores de la inflamación implicados en la artrosis
Los condrocitos presentan la capacidad de producir gran variedad de mediadores de la inflamación: proteasas (colagenasas, estromelisinas, agrecanasas), citocinas proinflamatorias (IL-1α, IL-1&abeta;, TNF-α, IL-8, IL-17, IL-18, proteína quimiotáctica para monocitos [MCP-1], RANTES), citocinas antiinflamatorias y antagonistas (IL-4, IL-10 e IL-13), factores de crecimiento (factor de crecimiento semejante a la insulina [IGF], factor transformador del crecimiento β [TGF-β]), radicales libres (NO) y mediadores lipídicos (prostaglandina E2 [PGE2], leucotrieno B4 [LTB4]) (tabla 1)18.

Metaloproteasas. El control del depósito de la matriz del cartílago no se realiza exclusivamente para la producción de los distintos componentes de la matriz extracelular, fundamentalmente proteoglicanos (PG) y colágeno tipo II, sino que es el resultado del equilibrio entre producción y degradación19. En condiciones normales, el condrocito mantiene un equilibrio estricto entre la síntesis y la destrucción de sustancias que produce habitualmente. En condiciones patológicas el condrocito responde a diversos estímulos con la producción de mediadores de la inflamación y enzimas, que alteran su metabolismo habitual. Como hemos comentado anteriormente, el rasgo principal de la OA es la destrucción progresiva de la matriz del cartílago (figs. 1 y 2). Esta destrucción de las moléculas de la matriz del cartílago implica la acción de unas enzimas proteolíticas denominadas metaloproteasas (MMP)20. Estas enzimas desempeñan un papel clave en la degradación irreversible de la arquitectura articular normal. Esta familia de enzimas se compone, al menos, de 18 miembros, que estructuralmente se clasifican en 5 subgrupos: a) colagenasas (MMP-1, MMP-8, MMP-13); b) gelatinasas (MMP-2, MMP-9); c) estromelisinas (MMP-3, MMP-10); d) estromelisinas de membrana (MMP-14, MMP-15), y e) otras (MMP-7, MMP-12). La actividad enzimática de estas enzimas está estrictamente controlada por sus inhibidores específicos, denominados inhibidores de tejido de MMP (TIMP). Se han identificado 3 formas diferentes de inhibidores de MMP en los tejidos de la articulación humana: TIMP-1, TIMP-2 y TIMP-3. Todos ellos se encuentran presentes en el cartílago y se sintetizan por los condrocitos OA. Las MMP se secretan en forma de proenzima inactiva y requieren ser activadas para poder ejercer su efecto biológico. En el cartílago artrósico, existe un desequilibrio entre la cantidad de TIMP y las MMP, y se produce una deficiencia relativa de los inhibidores. Se ha observado un desequilibrio similar entre la concentración de MMP y los TIMP en el líquido sinovial de la OA y en la expresión del tejido sinovial artrósico21. En la actualidad, conocemos que las colagenasas, las estromelisinas y las gelatinasas son enzimas determinantes en el proceso de destrucción del cartílago articular. Los valores de colagenasa y también de estromelisina están aumentados en el cartílago artrósico y guardan relación con la gravedad de las lesiones de OA en el cartílago artrósico22. Asimismo, se ha demos trado recientemente que los valores de MMP-3 (estromelisina-1) en el fluido sinovial de pacientes con OA, son extremadamente altos comparados con los de otras MMP23. La importancia de esta enzima se debe no sólo a su capacidad para degradar múltiples componentes de la matriz extracelular, sino por su capacidad de activar formas inactivas de otras MMP. Por ello, MMP-3 puede tener un papel central en la iniciación y en la progresión de la destrucción de la matriz del cartílago24. Otras enzimas que pueden desempeñar un papel fundamental en la destrucción del cartílago son la MMP-7 y MMP-13 ya que únicamente se han detectado en cantidades importantes en el cartílago OA y no en la membrana sinovial OA o AR25,26.
Aunque hasta el momento han sido las MMP las principales enzimas involucradas en la destrucción de la matriz del cartílago, no se puede descartar la participación de otros grupos de enzimas como los activadores del plasminógeno/plasmina y el grupo de las catepsinas. Las primeras son capaces de activar la colagenasa. En este sentido, los valores de plasmina y de colagenasa presentan una correlación directa y positiva en el cartílago artrósico. Además, el valor del inhibidor del activador del plasminógeno se ve disminuido en el cartílago de la OA27. Las segundas producen la degradación del colágeno y de los PG, al tiempo que activan diferentes MMP. En el cartílago de la OA, el valor de la catepsina B aumenta, y disminuye la actividad de su proteína inhibidora28. Por último, no podemos olvidar la actividad agrecanasa, otra proteasa que actúa rompiendo un enlace entre los dominios G1 y G2 del agrecano. Los valores de esta enzima pueden ser inducidos por la IL-1β. No se conoce su función específica en la OA, pero en el líquido sinovial de pacientes con OA se han encontrado fragmentos de agrecanos que se habían roto por el punto exacto en el que actúa esta enzima29.
Finalmente, destacar que la excesiva degradación de los principales componentes de la matriz extracelular del cartílago, como son el colágeno tipo II y el agrecano, puede representar un estado de la evolución de la OA relativamente tardío. Los episodios iniciales, que resultan de la hidratación del cartílago y del cloning de los condrocitos puede involucrar la degradación de otros componentes de la matriz, como pequeños PG y colágenos minoritarios, que son necesarios para la integridad y estabilidad de la matriz. Las proteinasas responsables de estos episodios pueden representar la diana terapeútica de intervención de esta enfermedad30.
Oxido nítrico. El NO es un mediador que está claramente involucrado en la destrucción del cartílago en la OA a través de diferentes mecanismos (fig. 1)2-13,31. El líquido sinovial procedente de las articulaciones humanas sanas contiene valores muy bajos de NO, a diferencia del procedente de pacientes con OA o AR que posee altas concentraciones de NO32. Además, la cantidad de NO producido por el tejido artrósico está en relación directa con el grado de lesión del cartílago33. De los tejidos que forman parte de la articulación, el cartílago es el tejido que tiene mayor capacidad para sintetizar y liberar NO. Los condrocitos superficiales estimulados con IL-1β producen más NO que las células de las capas más profundas34. En cambio, el tejido sinovial humano tiene una escasa capacidad para sintetizar y liberar NO35. La producción del NO se realiza a partir de la oxidación del aminoácido L-arginina por una familia de enzimas llamadas sintetasas del NO (NOS). Se conocen 3 isoformas, 2 de éstas se expresan de forma constitutiva (cNOS) localizada en neuronas (nNOS) y endotelio (eNOS) y una tercera isoforma inducible (iNOS)36,37.
El NO puede inhibir la síntesis de macromoléculas de la matriz del cartílago, tales como agrecanos. Asimismo, induce la inhibición de la actividad TGF-β38. También puede aumentar la actividad de MMP, incrementar la susceptibilidad al daño inducido por otros oxidantes (H2O2), reducir la síntesis del antagonista del receptor de IL-1 (IL-1Ra), inhibir la proliferación e inducir apoptosis10,39-42. Por otro lado, recientemente se ha implicado al NO en la formación de osteofitos43. Asimismo, se ha observado que el NO puede inducir la mineralización del cartílago en OA por medio de la inhibición de la fosforilación oxidativa mitocondrial44. Por ello, debido a las propiedades eminentemente destructivas del cartílago articular, el NO puede ser un importante mediador de la lesión articular crónica en la OA. En esta línea se ha demostrado que los condrocitos artrósicos expresan iNOS (ARNm y proteína) y producen NO de forma espontánea45. Sin embargo, el cartílago articular normal no produce NO ni contiene expresión alguna de la iNOS, a menos que se estimule con citocinas proinflamatorias como son IL-1β y TNF-α. El cartílago artrósico expresa iNOS y produce nitritos después de 72 h en cultivo, en ausencia de estímulos como IL-1β y TNF-α. Esto indica que los estímulos existentes en la matriz extracelular del cartílago artrósico persisten durante un tiempo y son, por tanto, capaces de inducir la síntesis de NO. Dichos estímulos podrían incluir: a) citocinas autocrinas o factores de crecimiento producidos por los condrocitos; b) interacción con componentes de la matriz extracelular (fibronectina), que incrementa la expresión de NOS, y c) difusión en la matriz de estímulos solubles desde otras células sinoviales46.
Por todo ello, la inhibición selectiva de iNOS puede proporcionar efectos positivos en el control de la evolución de la OA. El conocimiento sobre los mecanismos moleculares y celulares que implican la producción de NO puede ayudar al entendimiento de la enfermedad articular y contribuir al desarrollo de nuevas estrategias terapéuticas.
Eicosanoides: prostaglandinas y leucotrienos. Los tejidos articulares artrósicos liberan gran cantidad de prostaglandinas, principalmente PGE2, importante mediador de la inflamación45. Las prostaglandinas intervienen tanto en los fenómenos inflamatorios como en los destructivos de la enfermedad. Los signos clásicos de la inflamación aguda (dolor, eritema, edema y calor) pueden ser reproducidos por las acciones de las prostaglandinas al estimular la vasodilatación, incrementar la permeabilidad vascular y contribuir a la sensibilización al dolor. Asimismo, la PGE2 puede desempeñar un papel clave como mediador de la erosión del cartílago y del hueso yuxtaarticular al incrementar la actividad de MMP de la matriz, importantes mediadores de la degradación tisular47. Además, la PGE2 estimula la producción del factor de crecimiento endotelial vascular contribuyendo a los fenómenos de angiogénesis48. La interacción entre células residentes y células infiltrantes estimula la producción local de prostaglandinas durante la inflamación. Los leucocitos que invaden los tejidos articulares contribuyen a la producción de prostaglandinas por mecanismos que incluyen el contacto directo célula-célula y, en particular, la producción de citocinas47.
La regulación en la producción de prostaglandinas involucra numerosos pasos enzimáticos. La ciclooxigenasa (COX) es la enzima esencial en la biosíntesis de las prostaglandinas45. Este sistema es activado por la fosfolipasa A2, que libera el ácido araquidónico, sustrato de la COX. De este modo, esta enzima aumenta la producción de prostaglandinas. Se conocen 2 isoformas, COX-1 constitutiva (responsable de las funciones fisiológicas) y COX-2 inducible (interviene en la inflamación). La regulación de la expresión de COX-2 es un factor determinante para la producción de prostaglandinas durante la inflamación.
La PGE2 es un importante mediador lipídico en el metabolismo del cartílago. La expresión de la forma inducible de la ciclooxigenasa, COX-2, está incrementada en explantes de cartílago de pacientes con OA, que espontáneamente liberan PGE245. En cambio, los condrocitos articulares procedentes de donantes sin ninguna enfermedad articular expresan únicamente COX-149,50. De este modo, la PGE2 producida por los condrocitos activados por la enfermedad OA pueden modular la progresión de la enfermedad.
Diversos trabajos demuestran la existencia de una estrecha relación entre los mediadores de la inflamación NO y la prostaglandinas que pueden regular la homeostasis del tejido y contribuir al proceso fisiopatológico51. Sin embargo, los resultados obtenidos en cuanto a la conexión entre el NO y la enzima de la ciclooxigenasa son muy contradictorios y parecen depender del tipo celular y del microambiente presente. En los condrocitos humanos normales el NO aumenta la síntesis de PGE2 y la PGE2 no tiene efecto en la producción de NO41. En cambio, la producción de NO por el cartílago OA inhibe la producción de PGE245. Esta variedad de efectos de las prostaglandinas puede ser consecuencia de la activación de diferentes receptores de prostaglandinas, al activar cada uno de ellos diferentes vías de señalización18.
Por último, la función que desempeñan los productos de la lipooxigenasa en el desarrollo de la OA no está clara. En el líquido sinovial de pacientes con OA se ha demostrado una elevada actividad LTB452. Asimismo, en la membrana sinovial de pacientes con OA se ha detectado tanto LTB4 como LTC4, pero no en el cartílago. Aunque los mecanismos de actuación de los leucotrienos no se conocen con exactitud, sí sabemos que LTB4 induce la producción de IL-1ß en sinoviocitos53. También, hallazgos recientes demuestran que el efecto de la presión sobre condrocitos en cultivo es capaz de inducir la producción de LTB4 y NO54.
Citocinas: IL-1βy TNF-α. Las citocinas son un grupo de mediadores solubles que están formados por proteínas de bajo peso molecular y cuya función es la comunicación intercelular. En el cartílago articular, las citocinas se han clasificado históricamente como catabólicas, anabólicas y moduladoras (tabla 1)55. En particular, las citocinas IL-1β y TNF-α se constituyen como unos de los principales mediadores en la destrucción del cartílago. Estas citocinas son capaces de inducir su propia producción, al tiempo que inducen en sinoviocitos y condrocitos la estimulación de otros mediadores como IL-8, IL-6, LIF (factor de inhibición leucocitaria), y de estimular la síntesis de proteasas, y PGE2.
La IL-1β es inicialmente sintetizase como un precursor, para ser liberada al medio extracelular en forma activa. Su activación depende de una proteasa denominada enzima de conversión de IL-1β (ICE), o caspasa-1. Esta enzima esta incrementada tanto en el cartílago como en la membrana sinovial OA56. La IL-1β es el prototipo de estímulo capaz de inducir respuestas catabólicas en el condrocito. Estimula la expresión de proteasas y disminuye la síntesis de TIMP57. Suprime la expresión del ARNm α 1 (II) del procolágeno, colágeno tipo II y IX, y la síntesis de proteoglicanos en los condrocitos58. En cambio, la IL-1 estimula la síntesis de colágeno I y III. Inhibe la hipertrofia de los condrocitos y la calcificación del cartílago59. También es un inhibidor potente de la proliferación del condrocito inducida por TGF-β41. Por último, también induce la síntesis de prostaglandinas y de NO a través de un incremento en las isoformas inducibles de las enzimas que los producen. La suma de todos estos efectos tiene como resultado final la reabsorción del cartílago, resultado que ha sido confirmado por los efectos de la inyección intraarticular de esta citocina60.
La IL-1 realiza diferentes efectos biológicos a través de su unión a receptores específicos (IL-1R). Se han identificado 2 receptores: IL-1R tipo I y tipo II61. El receptor tipo I se encuentra incrementado tanto en condrocitos como en sinoviocitos OA, lo cual hace a estas células más sensibles a la IL-1β62. Los 2 tipos de receptores se pueden escindir de la membrana celular y encontrarse en forma soluble (sIL-1R). De esta forma el receptor puede anular la función de la IL-163. Un antagonista natural de la IL-1 es IL-1Ra, que neutraliza la acción de la IL-1, y cuya expresión está elevada en el daño articular y su síntesis está incrementada por los condrocitos artrósicos64. Diversos estudios in vivo, que emplean inyecciones intraarticulares de este receptor, demuestran como disminuye la progresión de la OA65.
El TNF-α también es una citocina fundamental en la destrucción de la matriz extracelular y en la estimulación de la inflamación sinovial. El TNF-α se produce como un precursor inactivo unido a la membrana. Es necesaria la actuación de la enzima de conversión del TNF-α (TACE) para su activación66. Recientemente, se ha demostrado que esta enzima se encuentra elevada en el cartílago de pacientes con OA67. La forma nativa del TNF-α es un polipéptido de 45 kDa, constituido por monómeros de 17 kDa que se unen por interacciones no covalentes formando trímeros. Se han descubierto 2 receptores distintos para el TNF-α, de 55 y 75 kDa, (TNFR55 y TNFR75). El receptor TNFR55 parece ser el más implicado en la actividad TNF en condrocitos y sinoviocitos artrósicos. Un aumento de la actividad de este receptor se ha descrito en estas células68.
También se ha demostrado en líquido sinovial de pacientes con OA la presencia de receptores solubles del TNF-α (sTNFR)68. Los 2 receptores solubles sTNFR55 y sTNFR75 se producen de forma espontánea por los sinoviocitos y condrocitos artrósicos69. El significado biológico de estos receptores depende de la concentración en la que se encuentren en los tejidos articulares. Pequeñas concentraciones pueden actuar estabilizando a la molécula del TNF-α, al aumentar su vida media. Sin embargo, concentraciones altas pueden reducir la actividad biológica del TNF al competir con los receptores de membrana por unirse al TNF-α. Por ello, los pequeños valores de sTNFR encontrados en el tejido pueden ser otro factor que favorezca los efectos catabólicos del TNF-α18.
Por último, basándose en el patrón de expresión de factores de crecimiento y citocinas observado en el cartílago articular de pacientes con OA, se han descrito recientemente 2 fenotipos diferentes de OA, denominados TNF-α bajo y TNF-α alto, que se caracterizan por un elevado número de condrocitos positivos para IL-1-β y TNF-α respectivamente. Estos fenotipos se han asociado con 2 genotipos diferentes. El fenotipo de TNF-α bajo se asocia con el alelo menos frecuente de IL-1-β y, el fenotipo de TNF-α alto se asocia con el alelo menos frecuente de IL-1Ra. Dependiendo de si los valores de IL-1-β o IL-1Ra están elevados, la patogería de la OA puede progresar de forma diferente conduciendo a los fenotipos de TNF-α bajo y TNF-α alto, respectivamente70.
Factores de crecimiento: IGF y TGF-β. Son proteínas que intervienen de forma decisiva en el metabolismo del cartílago. Generalmente tienen un efecto anabólico en el cartílago articular. De todos los facto res de crecimiento, los que más se han estudiado en el cartílago son el IGF-I y el TGF-β.
En el contexto de la biología articular, el IGF-I es el principal candidato responsable del anabolismo de la MEC del cartílago. Así, se ha visto que el IGF-I desempeña un papel fundamental en la regulación del metabolismo de proteoglicanos por el condrocito71. Aunque la expresión y síntesis de IGF-I están incrementadas en el cartílago OA, los condrocitos presentan una hiporrespuesta a este factor. Este fenómeno parece estar relacionado, al menos en parte, con un incremento en los valores de la proteína de unión a IGF (IGFBP), que controla la actividad biológica de IGF72. Asimismo, se ha demostrado que el NO es capaz de inhibir la respuesta de los condrocitos a IGF-I a través de la inhibición de la fosforilación de su receptor, el IGF-IR73. El TGF-β estimula la síntesis de proteoglicanos y la formación de osteofitos. También induce la síntesis de colagenasa-3 en condrocitos OA74.
Influencia de los estímulos mecánicos en la fisiopatología del condrocito
Entre los distintos factores que últimamente se han identificado como responsables del inicio y/o evolución de la OA, cabe destacar los factores mecánicos4. En este sentido, existe una relación directa entre la sobrecarga a la que está sometida una articulación y la aparición de la artrosis3. Sin embargo, dado que el cartílago hialino articular maduro es un tejido avascular, la nutrición de este tejido ocurre mediante difusión simple desde el hueso subcondral o la superficie articular. Por esta razón, la aplicación de la presión correcta sobre el cartílago articular es necesaria para su perfecta nutrición. Por ello, tanto el exceso como el defecto de presión se correlaciona con un metabolismo catabólico del cartílago. En este sentido, existen algunas publicaciones en las que se muestra que la presión aplicada sobre el cartílago es capaz de influir en la secreción de proteoglicanos, colágeno, citocinas, PGE2, NO y MMP75-77. En concreto, se demuestra la existencia de una relación directa entre el estrés mecánico y la inflamación del cartílago. En este sentido, se ha visto como la presión es capaz de inducir la producción de LTB4 y NO en condrocitos en cultivo54. En relación con las MMP, se ha visto como la presión es capaz de inducir la estimulación de MMP-9 a través de la activación del factor de transcripción c-jun77. Es de destacar, que tanto el exceso de presiones aplicadas sobre este tejido como la inmovilización de una articulación reduce la síntesis de proteoglicanos por los condrocitos75. En las células, la compresión del cartílago articular produce deformación de los condrocitos. Asimismo, se ha comprobado que las compresiones sobre el condrocito se acompañan de variaciones en los gradientes de presión hidrostática, flujo de fluidos, potenciales de membrana así como de cambios fisicoquímicos, como en el contenido de agua de la matriz, variaciones de la concentración iónica y cambios en la presión osmótica78. En relación con estas variaciones, cabría comentar que en la artrosis precoz el contenido intracelular de Na+ y K+ se mantiene en concentraciones similares a la de los condrocitos sanos; sin embargo, cuando la artrosis avanza la concentración intracelular, principalmente de Na+, aumenta de forma significativa. Además estos cambios en la concentración de Na+ dan lugar a variaciones en la síntesis de la matriz extracelular del condrocito79.
Cristales y osteoartritis
Actualmente se acepta que la inflamación es un componente más en el curso de la OA. Los cristales son uno de los muchos factores responsables de la inflamación presente en la articulación con OA. Los cristales más frecuentes en OA son los de pirofosfato cálcico y los de hidroxiapatita80. Recientemente, se ha demostrado que la IL-1 induce la promineralización del cartílago, al promover el depósito de los cristales de apatita, a través de un incremento de la actividad de 2 transglutaminasas, el factor XIIIa y la transglutaminasa de tejido. La actividad de estas enzimas en el cartílago articular se eleva con la edad y en los procesos degenerativos81.
Los cristales pueden causar la degeneración del tejido articular de una forma directa o paracrina. En el primer caso, los cristales inducen directamente a los sinoviocitos a proliferar, producir metaloproteasas y prostaglandinas. En el segundo caso, los cristales interaccionarían con los macrófagos/monocitos, con la consecuente liberación de citocinas que reforzarían la acción de los cristales sobre los sinoviocitos e inducirían a los condrocitos a la secreción de enzimas, que podrían favorecer la degradación de los tejidos articulares82,83.
La membrana sinovial en la artrosis
Hasta hace poco se consideraba que la afectación del tejido sinovial en la artrosis era mínima y secundaria al daño originado en el cartílago articular. Ahora se sabe que en las fases finales de la artrosis, la membrana sinovial desarrolla una respuesta inflamatoria que contribuye de manera decisiva en la patogenia y en el grado de expresividad clínica de la enfermedad84. Los cambios patológicos que se producen en la membrana sinovial de un paciente con un grado severo de OA son próximos a los cambios observados en la membrana sinovial de un paciente con AR6. En ambos casos se puede observar proliferación de las células sinoviales residentes y el acúmulo de una variada población de células inflamatorias, incluyendo células B y T activadas, en la membrana y el líquido sinovial. También se han descrito diversos grados de sinovitis en estadios tempranos de la enfermedad85. La inflamación sinovial está claramente reflejada en muchos de los signos y síntomas de la OA tales como: calor, enrojecimiento, edema e hinchazón18.
Las principales observaciones que sugieren que existe una asociación entre la inflamación y la progresión de los cambios estructurales en la OA, han surgido de diversos estudios clínicos18. Gran parte de estos estudios han demostrado una posible e interesante asociación entre sinovitis, inflamación OA y progresión de los cambios estructurales86,87. En este sentido, la presencia de la inflamación sinovial en la OA puede ser de suma importancia en el proceso de cronicidad de la degeneración articular, dado que la membrana sinovial activada sintetiza y libera diversos mediadores de la inflamación, como pueden ser las citocinas proinflamatorias (IL-1α, IL-1β, TNF-α, IL-8, MCP-1), proteasas (colagenasas, estromelisinas, agrecanasas), mediadores lipídicos (PGE2, LTB4) y radicales libres (NO, O_) que pueden modular el metabolismo condrocitario (tabla 1)88-90. De esta forma, se favorece la destrucción del cartílago y, al mismo tiempo, se estimula la síntesis de más mediadores proinflamatorios por el condrocito, originándose así un círculo cerrado que conduce a la destrucción de la matriz extracelular del cartílago hialino articular. En este sentido, diversos grupos han demostrado que las citocinas, particularmente la IL-1β, estimulan los condrocitos para liberar proteasas e inhibir la producción de la matriz cartilaginosa91. Por otro lado, la IL-1β también es capaz de inducir, en condrocitos procedentes de donantes sanos la producción de la quimiocina RANTES y de su receptor. Esta quimiocina y su receptor se encuentran elevados en la OA. El tratamiento del cartílago articular normal con RANTES da lugar a un incremento en los valores de NO, IL-6, MMP-1, aumenta la liberación de glicosaminoglicanos y disminuye profundamente la intensidad de la tinción de proteoglicanos con O-safranina92. Asimismo, se ha demostrado que en el tejido sinovial de pacientes artrósicos existen concrentraciones elevadas de estromelisina y de colagenasa93 y que estas concentraciones influyen directamente en la severidad de la inflamación y al mismo tiempo están relacionados positivamente con el nivel de IL-1β en el líquido sinovial. Dado que el tejido sinovial libera IL-1β, es probable que se produzca estimulación autocrina de la síntesis de las MMP por la membrana sinovial84. Como hemos comentado anteriormente la IL-1 ejerce su actividad biológica a través de la unión a sus receptores IL-1R I y II. Se ha demostrado que el número de receptores IL-1R tipo I está significativamente incrementado en la membrana sinovial de pacientes con OA62. Sin embargo, la membrana sinovial de un paciente con OA no produce solamente mediadores proinflamatorios. En este sentido, la acción de IL-1 puede ser modulada por el ya mencionado antagonista del receptor de la IL-1. El tejido sinovial artrósico expresa niveles elevados del gen y de la proteína de este antagonista del receptor94. Asimismo, la membrana sinovial OA es capaz de sintetizar IL-4, IL-10, e IL-13. Estas citocinas se encuentran incrementadas en el líquido sinovial de pacientes con OA y el resultado de sus efectos antiinflamatorios se traduce en una disminución de la producción de IL-1β, TNF-α y MMP, inhibición de PGE2, y un incremento en los valores de TIMP-1 y de IL-1Ra88,94. El tejido sinovial también es capaz de sintetizar IL-6, que podría tener un papel en el control del feedback negativo. Por otro lado, IL-6 es un importante factor responsable de la destrucción del hueso95.
Por último, no podemos olvidar que en los estadios finales de la OA, la inflamación sinovial se mantiene, al menos en parte, por fragmentos de cartílago articular y cristales de pirofosfato cálcico, de hidroxiapatita y de urato monosódico liberados del cartílago dañado. Estos factores son capaces de estimular las células de la membrana sinovial para sintetizar y liberar un elevado número de factores de la inflamación, que se asemejan a aquellos que se encuentran en enfermedades articulares inflamatorias tales como la AR80.
El hueso subcondral en la artrosis
La participación del hueso subcondral en la etiopatogenia de la OA es indudable. Incluso algunas teorías defienden que la OA es una enfermedad ósea más que cartilaginosa. En cualquier caso, la OA se puede definir como el proceso de degradación y pérdida del cartílago articular, acompañado por cambios hipertróficos del hueso con la formación de osteofitos y rigidez del hueso subcondral.
Uno de los posibles mecanismos de iniciación de la OA es la rigidez del hueso subcondral. Una vez iniciado el daño en el cartílago, la rigidez del hueso subcondral puede contribuir a una progresión más rápida de la OA96. El espesor y la densidad del hueso subcondral varía con la enfermedad articular. En una articulación normal, el hueso subcondral atenúa las cargas recibidas al absorber entre un 30-50% de la carga, mientras que el cartílago únicamente absorbe un 1-3%. Cuando el hueso subcondral se esclerosa, disminuye su capacidad de absorción hasta un 50% y ello supone un aumento de la energía que disipa al resto de la articulación, incluido el cartílago con el consiguiente deterioro de éste. Así, se han visto trabajos en los que alteraciones en el hueso subcondral preceden a los cambios en el cartílago97. El incremento en la rigidez del hueso subcondral puede ser parte de una alteración ósea más generalizada que conduce a un aumento en la mineralización y/o volumen de la articulación afectada. En este sentido, hemos de destacar que la OA no es frecuente en pacientes con osteoporosis, que presentan una disminución de la mineralización; mientras que la osteopetrosis, asociada a la esclerosis del hueso sí está relacionada con la OA. Estudios clínicos han demostrado que estas enfermedades, OA y osteoporosis, raramente se presentan juntas y que la presencia de una protege de la aparición de la otra98. Además, los pacientes con OA tienen, de forma significativa más hueso, independientemente del peso corporal, lo que sugiere que la OA puede, inicialmente, ser una enfermedad ósea. En esta línea de investigación, se han encontrado valores más elevados de hormona del crecimiento en pacientes con OA, en relación con aquellos que tienen osteoporosis. La hormona del crecimiento estimula la síntesis de IGF-I y ésta puede ser la razón de los elevados valores de IGF que se han encontrado en la cresta ilíaca de sujetos con OA en las manos. La presencia de estos valores elevados de factores de crecimiento en zonas óseas distintas y alejadas de la articulación dañada, sugiere que las personas que desarrollan OA pueden tener una predisposición a la formación de hueso más denso y rígido que el hueso normal. En este sentido, recientemente se ha observado que los osteoblastos del hueso subcondral presentan un fenotipo alterado y que ello puede contribuir al desarrollo de la OA. En concreto, se ha visto que los osteoblastos de la zona dañada presentan mayor actividad de fosfatasa alcalina, mayor producción de osteocalcina, de TGF-β y de IGF-I e IGF-II y también de colágeno tipo I y MMP-2. Lo que supone un incremento en el metabolismo de los osteoblastos en la artrosis. Estas alteraciones fenotípicas de los osteoblastos en la OA pueden obedecer a un defecto intrínseco de las células o bien ser una respuesta secundaria a diversos factores locales o sistémicos99.
La IGF-I y la PGE2 son capaces de disminuir los valores del receptor de la hormona paratiroidea (PTH-R) en osteoblastos procedentes de pacientes con OA, esto puede favorecer la esclerosis del hueso subcondral vía inhibición del catabolismo dependiente de PTH100. La actividad biológica de IGF está controlada por la IGFBP. Una elevada actividad proteolítica dirigida contra esta proteína puede desencadenar la ruptura del equilibrio IGF-I/IGFBP y contribuir a la esclerosis del hueso subcondral101.
Además, un trabajo reciente demuestra que este incremento en el metabolismo de los osteoblastos esta polarizado dentro de la cabeza femoral, y es más activo en el compartimiento proximal que en el distal. Esta polarización observada en el tejido OA, no sé encontró en las cabezas femorales de control. Esta diferencia en el ciclo metabólico entre la zona proximal y la distal de la cabeza femoral, puede ser la consecuencia de la deformación articular que se observa en los pacientes con OA de cadera, ya que es necesaria una estrecha coordinación en la remodelación ósea para mantener la forma y la morfología del tejido. Además, esta deformación ósea puede exacerbar el proceso de la enfermedad, al crear nuevas sobrecargas mecánicas102.
El hueso subcondral está muy vascularizado, aunque muchos de los vasos no alcanzan el cartílago calcificado y, excepto en la enfermedad, no penetran en el cartílago articular. Los valores de factores angiogénicos en el líquido sinovial están incrementados sobre 2 veces en todos los pacientes con OA. Un fallo de los condrocitos al producir una concentración suficiente de inhibidores de proteasas, que pudiese prevenir la invasión vascular, también se ha propuesto como un factor patogénico de la OA103, pero todavía hoy, no se han establecido las causas y el efecto. Aunque la remodelación puede ser un hecho beneficioso en la OA al incrementar el área de contacto en la articulación y de esta forma reducir las fuerzas sobre el cartílago, comúnmente se acepta que la invasión vascular sobre el cartílago calcificado es un componente crítico en la invasión de la OA.
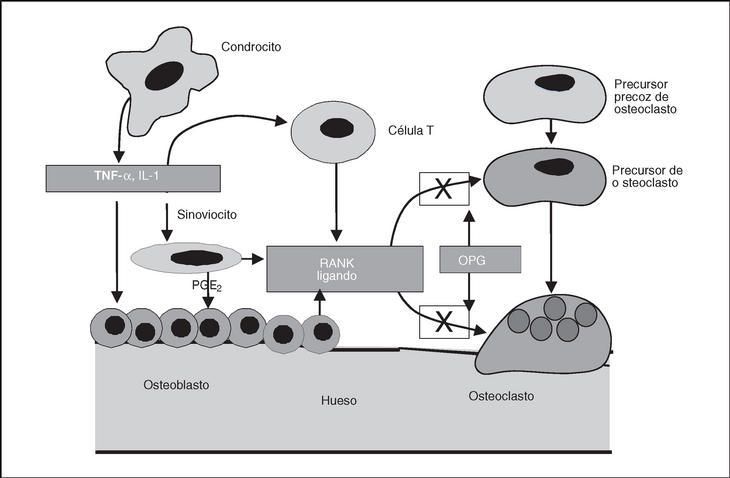
Recientemente, se ha descrito la participación de un nuevo sistema en la modulación del metabolismo óseo. Este sistema está formado por el ligando del receptor activador del factor nuclear αB (RANKL), su receptor, receptor activador del factor nuclear αB (RANK) y un antagonista del receptor denominado osteoprotegerina (OPG) (fig. 3). Así, se ha descrito cómo una actividad incrementada de RANKL provoca la reabsorción ósea. En cambio, la depleción de este factor conduce a una situación de osteopetrosis. La OPG antagoniza la acción de RANKL al inducir apoptosis de los osteoclastos e inhibir de esta manera la reabsorción ósea. Además, se ha visto que la OPG tiene efectos protectores sobre la matriz del cartílago mediante mecanismos distintos al bloqueo de RANKL104. En el líquido sinovial de pacientes con OA de rodilla se ha observado un incremento en los valores de OPG y ello podría reflejar una respuesta compensatoria a la degeneración del cartílago articular y servir de protección del cartílago más que representar una causa de OA105.
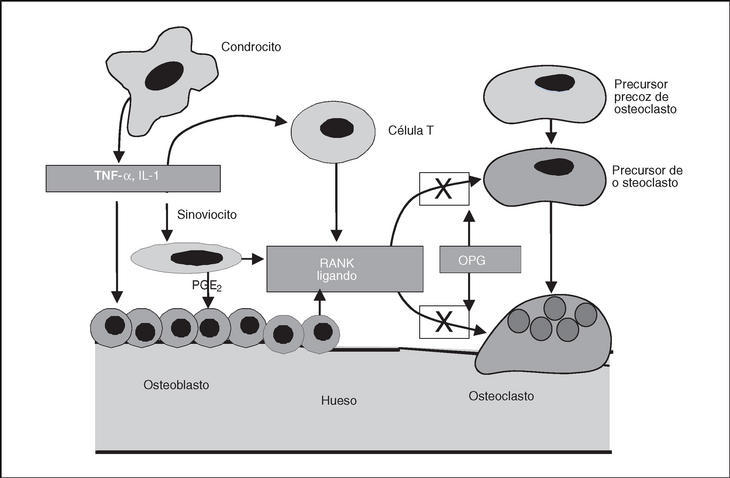
Figura 3. Mecanismo de acción de la osteoprotegerina (OPG) en el hueso subcondral. OPG: osteoprotegerina; PGE: prostaglandina-E2; RANK: receptor-activador.
Marcadores biológicos de la artrosis
En la articulación enferma, se produce una pérdida del balance normal entre síntesis y degradación de las macromoléculas que proporcionan, a los distintos tejidos que la componen, sus propiedades funcionales y biomecánicas. Esta alteración afecta a cada uno de los compartimientos que forman la articulación; es decir cartílago, hueso y membrana sinovial106. Como resultado de este metabolismo alterado, diferentes macromoléculas difundirán, en un primer momento, al líquido sinovial para, posteriormente, ser modificadas en el torrente linfático y llegar a la sangre que actuará como transportadora de los fragmentos hasta el hígado y el riñón, donde se fragmentan nuevamente y se eliminan en la orina. Fluidos biológicos en los que se pueden identificar y cuantificar. Estos marcadores bioquímicos pueden reflejar anormalidades en los distintos tejidos articulares y pueden ser útiles en la investigación y seguimiento de la OA106. Dentro de éstos podremos obtener marcadores que reflejen la actividad ósea, la sinovial y/o también la del cartílago. Éstos a su vez, pueden ser marcadores de síntesis o de degradación. Generalmente, se acepta que una alta actividad de la enfermedad sugiere una rápida progresión de la enfermedad.
Sin embargo, y a pesar de los numerosos esfuerzos realizados en los últimos años, realmente no se ha podido definir un marcador biológico que nos permita conocer el estado del cartílago en los distintos estadios de la enfermedad articular, predecir la progresión de la OA o evaluar el efecto de los distintos tratamientos empleados en ella107.
El sistema nervioso y el músculo en la fisiopatología de la artrosis
En la actualidad, la importancia del sistema neuromuscular en el inicio y la progresión de la OA no se conoce con profundidad. Por ello, hasta hace relativamente pocos años se pensaba que las terminaciones nerviosas presentes en la articulación, únicamente prestaban una función de transmisión de las sensaciones, tanto nociceptivas como propioceptivas, al sistema nervioso central (SNC). Actualmente sabemos que el dolor que está asociado con la OA puede originarse en el SNC, más que en la articulación o tejido periarticular, y que el SNC puede participar de una manera activa en la patogenia de la enfermedad108.
Las terminaciones nerviosas aferentes pueden contribuir al desarrollo de la enfermedad articular a través de una excesiva liberación en los tejidos articulares de neuropéptidos y otros mediadores de la inflamación109. Este proceso supone un incremento de la presión intraarticular que, presumiblemente, puede afectar a la neurofisiología de la articulación, dando lugar a una hiperexcitabilidad en estas terminaciones nerviosas que, finalmente, conlleva dolor articular. Esta hiperexcitabilidad de las terminaciones nerviosas aferentes conduce a una hiperexcitabilidad de las neuronas supraespinales y espinales, estableciéndose un mecanismo por el cual el dolor articular puede no depender, de forma directa, de los estímulos originales110.
Otra función de las terminaciones nerviosas aferentes en la articulación es transmitir información al SNC para que pueda ser utilizada en la coordinación de la actividad muscular y, de esta forma, proteger a la articulación de aquellos movimientos potencialmente peligrosos. Por ello, una pérdida de mecanorreceptores y la consecuente reducción de las aferencias neuronales, puede reducir la estabilidad funcional de la articulación, participando en los procesos degenerativos de la OA. De forma similar, las lesiones del SNC o del sistema periférico que afectan a la coordinación muscular también pueden iniciar o potenciar la lesión articular111.
El músculo tiene 2 funciones principales en la articulación. En primer lugar, proporciona la energía necesaria para que pueda desarrollarse el movimiento y, en segundo lugar, junto con otras estructuras intraarticulares y periarticulares, actúa proporcionando estabilidad a la articulación112. Por esta razón, una disminución importante de la fuerza muscular puede precipitar la degeneración de la articulación OA. Por ello, el músculo podría proporcionar un frente terapéutico interesante en el tratamiento de la OA. En este sentido, recientemente se ha publicado cómo un entrenamiento moderado supone mejoras sustanciales con relación a la fuerza, el dolor, la función física y la calidad de vida en los pacientes con una OA de rodilla113.
Agradecimientos
Los autores quieren manifestar su agradecimiento a la Srta. Pilar Cal Purriña por la ayuda prestada en la redacción de este documento.