


Caso clínico
Varón de 50 años de edad, fumador de un paquete/día desde los 18 años. Consumo de alcohol no relevante. Entre sus antecedentes patológicos figuran una exéresis de pólipos en las cuerdas vocales y de un carcinoma basocelular en la zona lumbar hace 2 años. Presenta un angioma plano congénito en la zona pectoral y un quiste epidermoide centrotorácico. A los 30 años fue diagnosticado en otro centro de una espondilitis anquilosante, con presencia de anquilosis lumbar y de las vértebras sacroilíacas asociado a HLA B27 positivo. Fue tratado con antiinflamatorios y glucocorticoides, y recibió radioterapia antiálgica lumbar. Posteriormente, su enfermedad reumatológica se mantuvo poco sintomática, por lo que no seguía ningún control ni tratamiento.
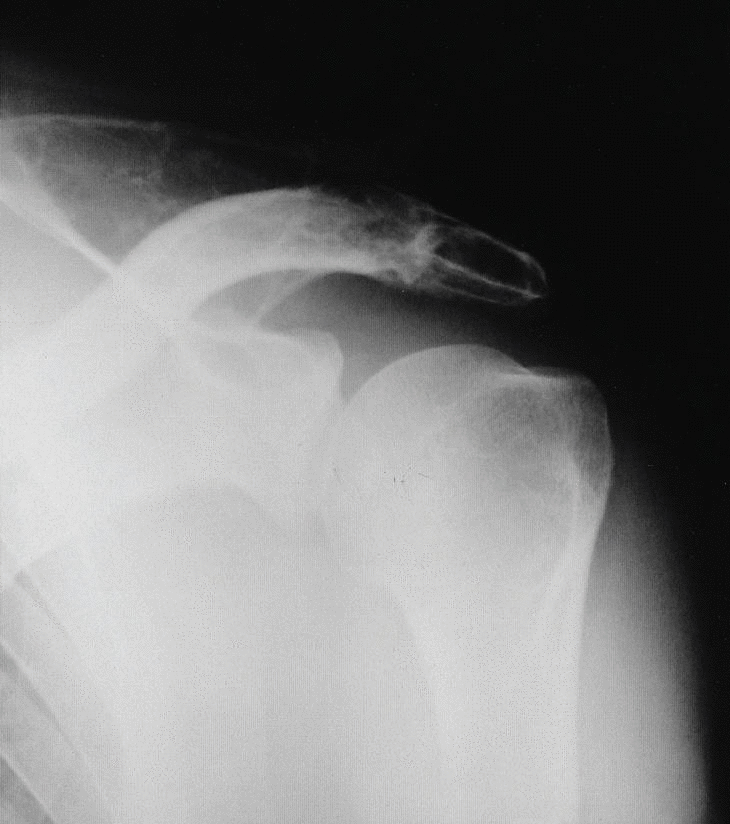
En julio de 2000 acude a su médico de cabecera a causa de un dolor en el hombro derecho de un mes de evolución, que se desencadenaba con los movimientos de la articulación y con el sobreesfuerzo de la extremidad. Se realizaron unas radiografías del hombro que fueron informadas como normales (no disponibles). Debido a la persistencia del dolor a pesar del tratamiento antiinflamatorio, fue remitido al reumatólogo de atención primaria. La exploración reumatológica en ese momento demostró la limitación axial propia de su enfermedad, con un test de Schober de 3 cm sin dolor a la presión vertebral o de sacroilíacas. La movilidad del hombro se hallaba conservada, aunque la abducción forzada era dolorosa. Se realizaron nuevas radiografías de hombro que objetivaron imágenes líticas en el acromion, la escápula y el húmero derecho, y una ecografía del hombro derecho que evidenció una lesión neoformativa que expandía e insuflaba el acromion y la escápula, rompiendo la cortical con infiltración de las partes blandas. La analítica en ese momento evidenció: VSG 10 mm en la primera hora (0-20); Hb 14,3 g/dl (13-18); Hto. 41,8% (39-54); VCM 94,3 fL (80-97); plaquetas 238 * 109/l (135-450 * 109); leucocitos 10,2 * 109/l (58,8% S, 32,6% L, 9,2% M, 5,3% E, 1,2% B); glucosa 5,1 mmol/l (3,6-6,4), uratos 327 µmol/l (202-482), colesterol 519 mmol/l (3,5-5,5); ALT 44 U/l (5-54); FFAA 217 U/l (78-279); calcio 2,3 mmol/l (2,1-2,6); fósforo 1,0 mmol/l (0,95-1,7); PCR 3,1 mg/l (0-5); sedimento de orina: normal.
El paciente fue ingresado en el servicio de reumatología de la CSUB para completar el estudio y la filiación de dicha lesión.
Al ingreso, el enfermo presentaba un buen estado general y se hallaba afebril, normocoloreado y normohidratado. No se hallaron adenopatías. Auscultación cardíaca rítmica y sin soplos. Auscultación respiratoria normal. Abdomen blando, no doloroso, no se palparon visceromegalias. Exploración neurológica dentro de la normalidad. Se apreció un discreto abombamiento de la zona acromial de la escápula derecha, con leve desplazamiento anterior del hombro, de consistencia normal. Dolor a la palpación del ángulo inferior escapular. No presentaba dolor a la movilización pasiva, aunque sí a la movilización activa extrema (abducción y rotación externa). Sin evidencia de linfedema. Fuerza y sensibilidad conservadas. Pulsos normales. Dolor a la palpación de los arcos costales 8-9 izquierdos en su plano posterior. Discretas molestias a la presión de los puntos sacroilíacos y de las apófisis espinosas lumbares. Limitación de la flexión lumbar sin variaciones respecto a la exploración anterior.
La analítica del ingreso evidenció: VSG 23 mm en la primera hora; Hb 13,8 g/dl; Hto. 39,8%; VCM 96 fL; plaquetas 238 *109/l; leucocitos 12,2 * 109/l (55% S, 32,1% L); glucosa 5 mmol/l (4,1-7,8), uratos 384 µmol/l (143-371), colesterol 5,2 mmol/l (2,93-5,2); ALT 1,02 µkat/l (N, 0,05-0,56); AST 0,43 µkat/l (N, 0,05-0,5); FFAA 1,8 µkat/l (N, 0,1-1,6); GGT 1,64 µkat/l (N, 0,05-0,53); bilirrubina 4 mmol/l (1-18); calcio 2,4 mmol/l (2,15-2,51); fósforo 1,4 mmol/l (0,85-1,5); Na 139 mEq/l (135-147); K 4,6 mEq/l (3,83-5,1); proteínas totales 61 g/l (64-80); PCR < 3 mg/l (0,1-5); fibrinógeno 4,6 g/l (2-4); tiempo de protrombina 0,89; TTPA 0,86; ferritina 214,6 µg/l (30-300); FR negativo; beta-2 microglobulina sérica 3,9 mg/l (N, 0,8-2,4 mg/l); IgG 477 mg/dl (N, 690-1.400); IgA 35,6 mg/dl (N, 88-410); IgM < 17 mg/dl (N, 34-210). Proteinograma: Alb 57,3% (N, 57-65); alfa-1 globulina 3,5% (N, 2-4); alfa-2 globulina 18,2% (N, 8-13); betaglobulina 14,7% (N, 9-14); gammaglobulina 6,3% (N, 10-19). Marcadores tumorales: CEA 3,5 µg/l (N, 0-34); AgCA 19,9 :15 karb.µ/l (N, 0-39); AgCA 125, 11,9 karb. µ/l (N, 0-35). Tira reactiva de orina normal.
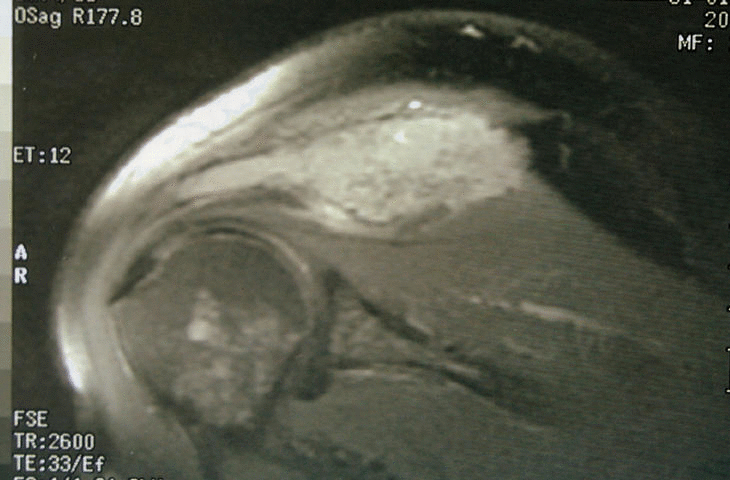
Radiografía de tórax: no se detectó la presencia de cardiomegalia, alteraciones pulmonares ni mediastínicas. Se aportan imágenes de la RM realizada en el hombro afectado.
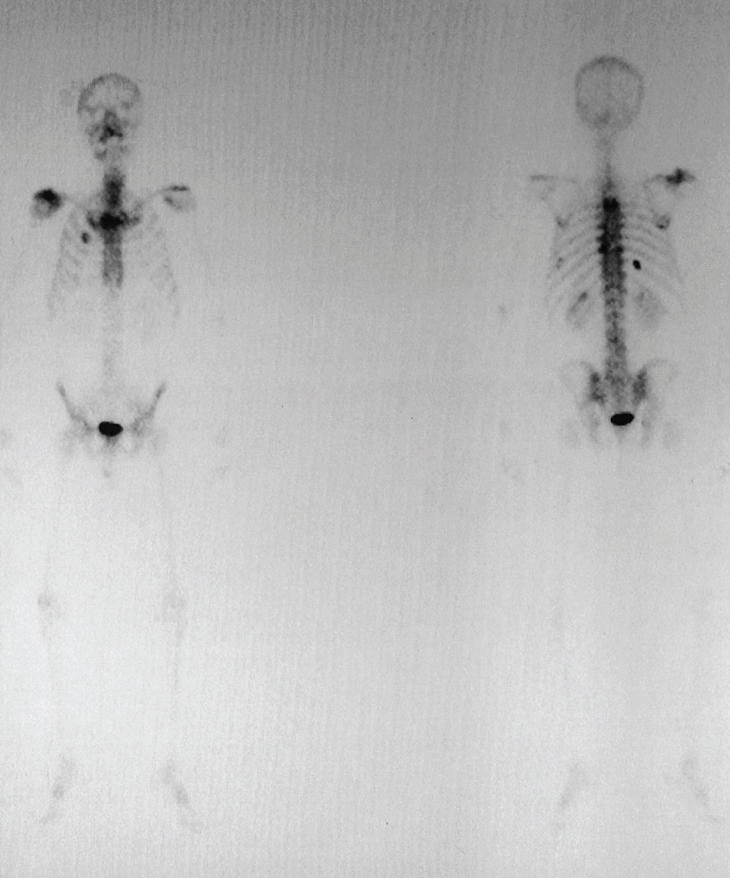
Gammagrafía ósea: proceso óseo activo sobre el hombro derecho. Acumulaciones en la cara anterior del segundo arco costal derecho, ambas regiones laterales de D3, articulación costovertebral izquierda de D9, octavo arco costal posterior izquierdo, décimo arco costal posterior derecho, punta de la escápula derecha y captación discreta en la región lateral derecha de D4.
Ecografía abdominal: sin alteraciones.
Se realizaron exploraciones que permitieron el diagnóstico.
Dra. Ortiz-Santamaría
Se trata de un varón de 50 años con antecedentes de espondilitis anquilosante tratada con radioterapia, y angioma plano congénito torácico, que presenta dolor, tumoración y limitación funcional del hombro derecho. A la radiografía simple se observan varias lesiones osteolíticas agresivas, en más de un hueso, de una misma región anatómica (fig. 1). En la gammagrafía ósea se objetiva un aumento de la captación del trazador en el acromion, así como en distintas costillas y la región lateral de D3 y D4 (fig. 2). En la RM del hombro se evidencia una lesión expansiva tumoral que reemplaza la médula ósea y destruye la cortical del omóplato, concretamente del acromion, con componente de partes blandas con efecto masa sobre los músculos supraespinoso y trapecio (fig. 3). Existen otras lesiones en el omóplato que también reemplazan la médula ósea y erosionan el endostio; también se observan en la epífisis del húmero y en los primeros arcos costales. Se trata de imágenes hipointensas en T1 e hiperintensas en T2. En el interior de la lesión situada en el acromion, se observan pequeñas imágenes de baja intensidad de señal, compatibles con áreas de calcificación o hemorragia. No existe compromiso articular. En la analítica destaca: leve linfomonocitosis, leve alteración de las enzimas hepáticas, disminución de las inmunoglobulinas, aumento de la alfa-2/beta globulina en el proteinograma, con leve aumento de la beta-2 microglobulina sérica. La uroanálisis elemental, la radiografía de tórax y la ecografía abdominal fueron normales.
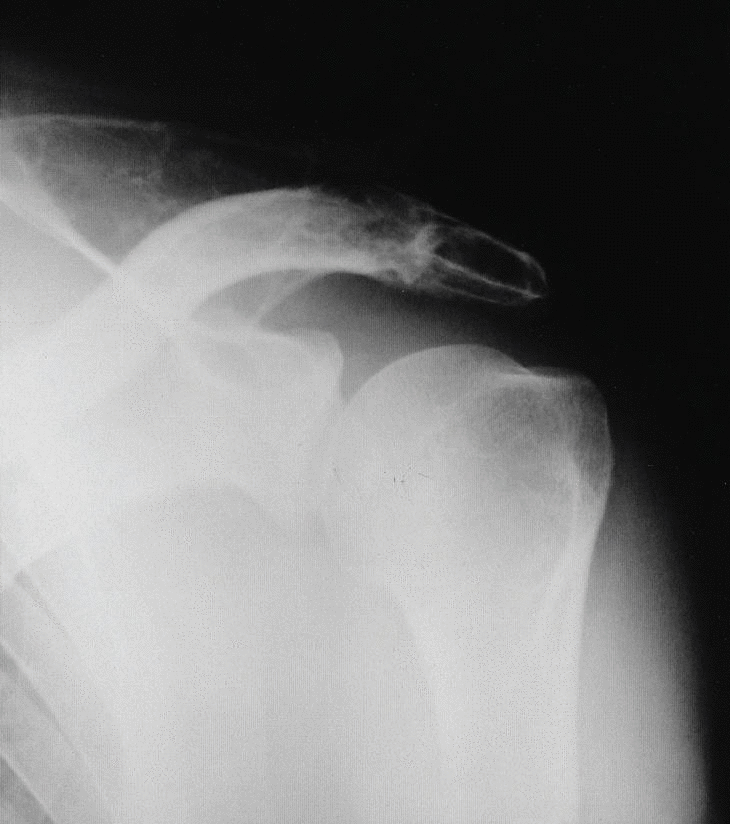
Figura 1. Radiografía simple de hombro: imágenes líticas en acromion, escápula y húmero
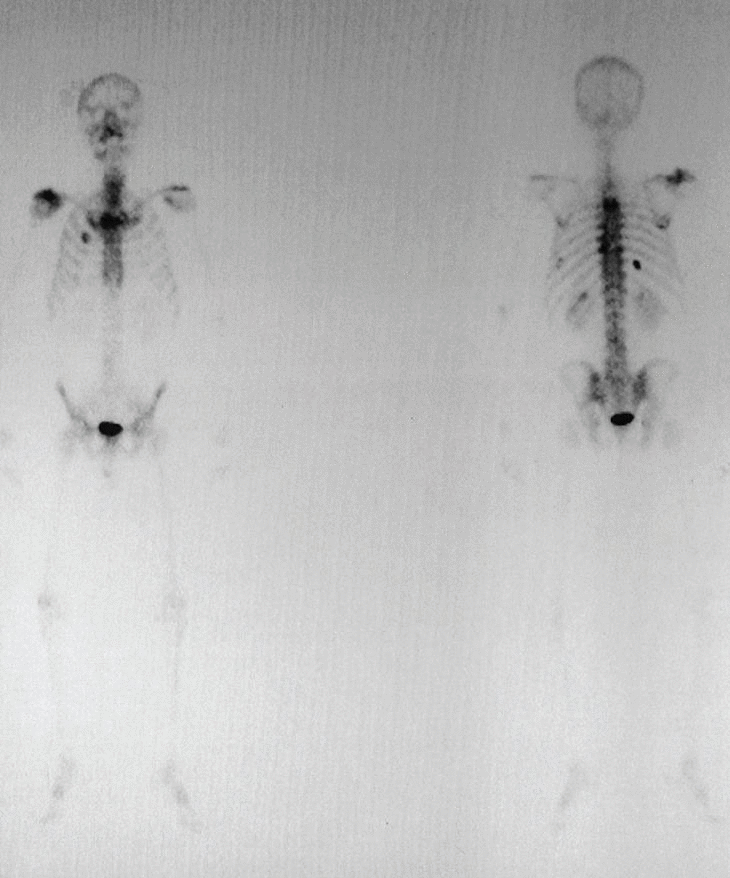
Figura 2. Gammagrafía ósea: aumento de la captación del trazador en el acromion, así como en distintas costillas y la región lateral de D3 y D4.
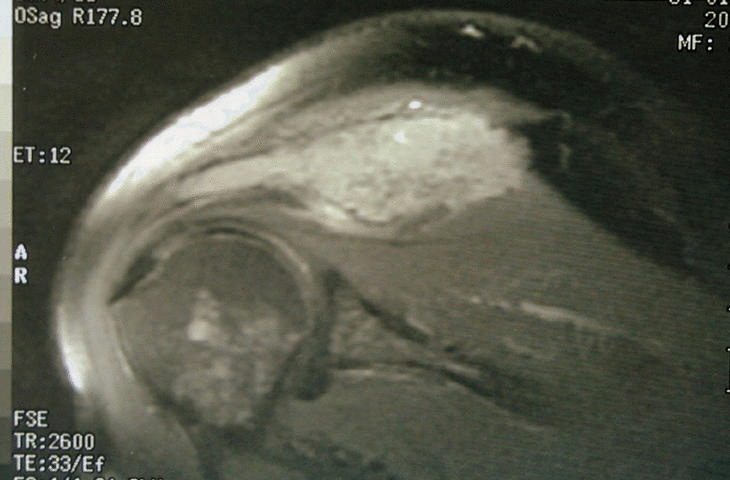
Figura 3. Resonancia magnética del hombro: lesión expansiva tumoral que reemplaza la médula ósea y destruye la cortical con componente de partes blandas.
Centraré el diagnóstico en un enfermo con espondilitis anquilosante y lesiones osteolíticas múltiples. En el caso de que estas lesiones osteolíticas estén en relación con la enfermedad de base, cabe destacar como complicación a largo plazo de la espondilitis anquilosante la aparición de una amiloidosis secundaria. La afección renal en forma de proteinuria y/o insuficiencia renal es la manifestación que alerta de la aparición de amiloidosis secundaria1-3; en cambio, la afección ósea de la amiloidosis secundaria es excepcional. A veces se observan depósitos tumorales de sustancia amiloide, llamados amiloidomas, generalmente del tipo AL (amiloidosis primaria), en los que no existe la evidencia de amiloidosis generalizada o de otra enfermedad sistémica4. También se han descrito casos de amiloidomas en tejidos blandos en los que la proteína amiloide era del tipo AA5 (amiloidosis secundaria). La localización más habitual de los amiloidomas es la columna vertebral y el cráneo, pero también el húmero, el omóplato y la pelvis. Son más habituales en varones de mediana edad (57 años) que en mujeres (2:1). La forma de presentación puede ser como afección neurológica (radiculopatía, paraparesia), tumoración dolorosa o fracturas patológicas. Radiológicamente, se caracterizan por lesiones líticas con destrucción de la cortical y masa de partes blandes con o sin áreas de calcificación, que pueden recordar un condrosarcoma6. El pronóstico está en relación con la posible progresión hacia una enfermedad sistémica, sea la amiloidosis o una hematopatía. Según Pambuccian et al6, los amiloidomas representarían plasmocitomas con depósito masivo de proteína amiloide, en donde la ausencia de componente monoclonal se explicaría por la baja solubilidad de las inmunoglobulinas con tendencia a la precipitación local. Otros autores consideran que el pronóstico de los amiloidomas es excelente sin progresión hacia enfermedades sistémicas ni recidivas4,7. En este caso, la presencia de afección ósea múltiple permite descartar el amiloidoma y la poca expresión biológica de la enfermedad, la existencia de reactantes de fase aguda poco elevados, la uroanálisis normal, aun sin conocer la función renal y la proteinuria, hacen poco probable el diagnóstico de amiloidosis secundaria.
En relación con el tratamiento recibido al inicio de la enfermedad, radioterapia antiálgica lumbar, se ha descrito un mayor riesgo de aparición de casos de leucemia, linfoma no hodgkiniano y mieloma múltiple, entre otros, respecto a los pacientes con espondilitis anquilosante no irradiados. Este riesgo disminuye con el paso de los años en relación con el momento del tratamiento radioterápico8,9. Según Cuzick et al10, la aparición de mieloma múltiple en los pacientes irradiados acontece después de 10 años de haber seguido el tratamiento y el riesgo puede continuar después de 30 años o más. Algún autor ha puesto en duda si la radioterapia es condición sine qua non, o bien la misma enfermedad lo podría favorecer11, en respuesta a un estímulo antigénico crónico12. Se ha descrito algún caso de espondilitis anquilosante asociada a mieloma no secretor13. Por orden de frecuencia, el mieloma múltiple afecta a las vértebras, las costillas, el cráneo, la pelvis y el fémur. En el caso del plasmocitoma término usado para las lesiones solitarias afecta a las vértebras, la pelvis, el cráneo, el esternón y las costillas. El mieloma representa el 1% de las neoplasias y el 10-15% de las neoplasias hematológicas. La edad del diagnóstico está entre los 40 y los 80 años y es más frecuente en los varones que en las mujeres. Clínicamente, se puede presentar en forma de afección neurológica, síndrome tóxico, fiebre, adenopatías, visceromegalias y fracturas patológicas, entre otros procesos. En la analítica a menudo destaca la presencia de anemia, reactantes de fase aguda elevados, alteraciones en el proteinograma, presentes en el 80-90% de los enfermos con lesiones óseas múltiples, en forma de componente monoclonal IgG, y puede existir proteinuria de Bence-Jones. Radiológicamente, el mieloma se caracteriza por lesiones líticas, de tamaño regular, expansivas, que se pueden acompañar de masa de partes blandas y pueden estar calcificadas, sobre todo cuando está asociado a amiloidosis (el 15% de los mielomas múltiples presentan depósitos de proteína amiloide). Clásicamente, la captación de la lesión en la gammagrafía ósea es de baja intensidad o inexistente, pero se han descrito casos con elevada intensidad de la captación12, en relación con fracturas patológicas o destrucción de la cortical. En este caso, la localización ósea múltiple, el aumento de la beta-2 microglobulina sérica y de los reactantes de fase aguda y la disminución de las inmunoglobulinas son datos que inclinan hacia el diagnóstico de mieloma, aun sin conocer la existencia de proteinuria ni de componente monoclonal. Otra hemopatía a tener en cuenta es el linfoma óseo. El linfoma óseo primario también es más frecuente en varones de más de 40 años, pero sólo representa el 7% de los tumores óseos malignos. Se localiza habitualmente en el fémur y la pelvis. Son lesiones únicas en el 66-80% de los casos y, cuando son múltiples, se trata de distintas lesiones en un mismo hueso o lesiones únicas en distintos huesos. Adoptan un patrón lítico (51%), esclerótico (42%) o mixto, con reacción perióstica leve y masa de partes blandas14-16. Los síntomas sistémicos son infrecuentes. Teniendo en cuenta que es una neoplasia poco frecuente y que no es la localización habitual del linfoma, considero este diagnóstico menos probable que el de mieloma.
Hay que tener en cuenta el diagnóstico de metástasis de origen desconocido. El dolor es el síntoma más habitual, a menudo producido por una expansión ósea o por una fractura patológica. Las neoplasias que con más frecuencia diseminan el hueso son las de próstata, mama, pulmón, tiroides y riñón; las últimas son las más osteolíticas. Las metástasis se localizan sobre todo en el esqueleto axial y pueden ser líticas o escleróticas. Pueden presentar una expansión ósea, pero la reacción perióstica y la masa de partes blandas es más frecuente en los tumores óseos primarios. La afección de la clavícula, el acromion, la glenoides y el olécranon es más típica del mieloma17. En este caso, la localización en el hombro, la ausencia de síndrome tóxico y la poca repercusión analítica hacen que el diagnóstico de metástasis no sea el primero a tener en cuenta.
El enfermo presenta un angioma plano congénito torácico, lo que plantea la posibilidad de un tumor vascular. La clasificación de los tumores óseos de origen vascular está extensamente discutida, pero se podría sintetizar en tres grandes grupos: los congénitos, como el angioma plano, los adquiridos, como el hemangioma cavernoso, y los malignos, como el angiosarcoma. La afección ósea en forma de lesiones múltiples en más de un hueso de la misma extremidad es muy característico de los tumores vasculares18,19. Se ha descrito la afección ósea simultáneamente a la cutánea20,21, en el caso de los tumores óseos vasculares adquiridos, que no siempre se corresponde a la misma metámera22. En la revisión realizada no se ha hallado ningún caso de afección ósea y cutánea en pacientes con tumores óseos vasculares congénitos, como el que presenta el caso expuesto. Las imágenes típicas por RM de los tumores vasculares son masas de vasos tortuosos y flebolitos en las partes blandas18,20, características que se pierden en caso de tumores óseos vasculares agresivos. Teniendo en cuenta la afección ósea múltiple, el aumento de la beta-2 microglobulina sérica y la disminución de las inmunoglobulinas hacen que este diagnóstico quede en segundo término.
Otras posibilidades, como la histiocitosis X y la displasia fibrosa poliostótica, son más habituales en gente más joven; para el diagnóstico de osteomielitis, el caso expuesto no presenta compromiso articular ni otras características radiológicas típicas23-25.
Con los argumentos expuestos, me inclinaría a realizar como pruebas diagnósticas una proteinuria de 24 h, en busca de un componente monoclonal, mediante inmunofijación en orina, o bien, en sangre, un aspirado de médula ósea y, si se requiere, una biopsia del acromion derecho, ya que la presunción diagnóstica es, en primer lugar, el mieloma múltiple, sin descartar que en el seno de la lesión se identifique proteína amiloide AL.
Dra. Lourdes Mateo
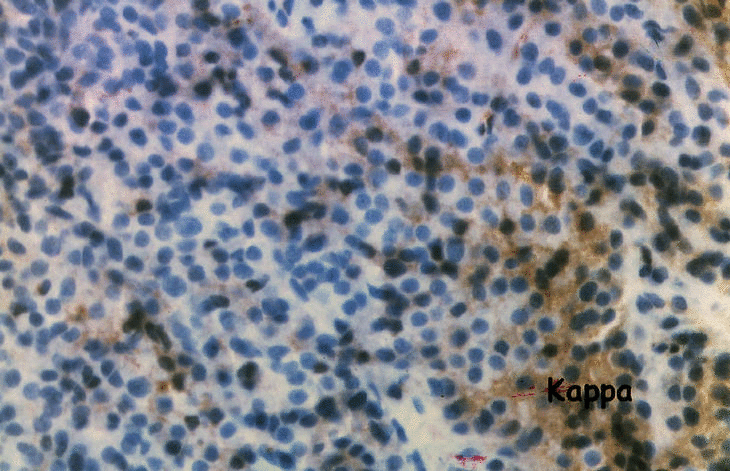
La primera maniobra diagnóstica realizada fue la punción-biopsia con thru-cut mediante control por escáner en la tumoración localizada en el acromion derecho. El estudio anatomopatológico demostró un infiltrado mayoritario de células plasmáticas. Las técnicas de inmunohistoquímica con anticuerpos contra cadenas lambda fueron positivas, en tanto que la tinción para cadenas kappa fue negativa (fig. 4a y b). El diagnóstico anatomopatológico fue de plasmocitoma monoclonal lambda.
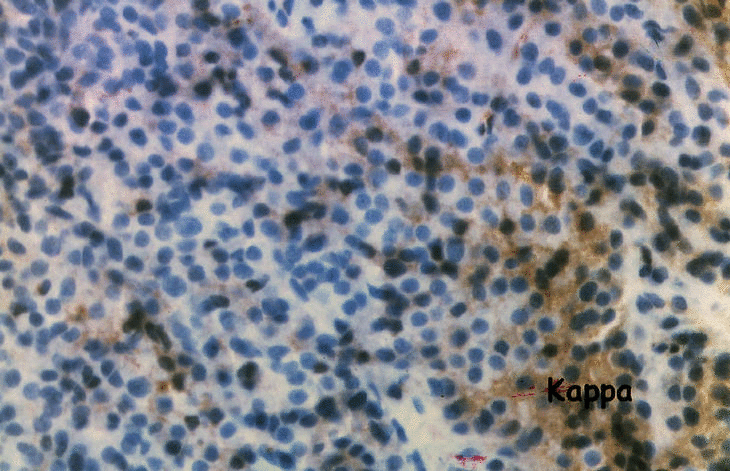
Figura 4. Punción-biopsia de la tumoración acromial: a) infiltrado de células plasmáticas con presencia de cadenas ligeras lambda monoclonales; b) la tinción de cadenas kappa es negativa.
La ausencia de banda en el proteinograma sérico, la ausencia de anemia, la normalidad de la calcemia y de la función renal podían orientar hacia un plasmocitoma multifocal, pero otros datos, como el notable descenso de todas las inmunoglobulinas y la elevación de la beta-2 microglobulina sérica sugerían el diagnóstico de mieloma múltiple.
Paralelamente a la punción-biopsia, el estudio de paraproteínas por técnicas de inmunofijación confirmó la presencia de cadenas ligeras lambda monoclonales, tanto en suero como en orina, con proteinuria de Bence-Jones.
Así pues, el diagnóstico definitivo fue de mieloma múltiple Bence-Jones de cadenas ligeras lambda, estadio IIIA.
A las 3 semanas del ingreso la radiología seriada ósea evidenció la existencia de múltiples lesiones líticas en la calota craneal, el acromion, el húmero derecho y el húmero izquierdo, y la ausencia de los pedículos vertebrales de L4 y L5. La RM de columna cervico-dorso-lumbar objetivó una infiltración en múltiples vértebras.
El paciente fue tratado con 6 ciclos de quimioterapia (vincristina 0,4 mg, farmiblastina 17 mg, dexametasona 40 mg) y pamidronato intravenoso para el tratamiento paliativo de las lesiones osteolíticas, con buena tolerancia. Posteriormente, se procedió a realizar un autotrasplante de médula, indicado por la edad del enfermo y el avanzado estadio del mieloma. La evolución a corto plazo ha sido satisfactoria y la detección actual de paraproteínas es negativa tanto en suero como en orina.
La supervivencia del autotrasplante en estos enfermos se sitúa en torno a los 50 meses, y a los 10 años la tasa de supervivencia actual es del 30%26. Los factores que sugieren un mejor pronóstico son la edad menos avanzada, la respuesta favorable a la quimioterapia, los estadios I y II del mieloma y las concentraciones bajas de beta-2 microglobulina sérica en el diagnóstico27.