






Caso clínico
Mujer de 27 años de edad, fumadora de 15 cigarros al día y sin antecedentes de alergia. En enero de 1994 inició artritis intermitente que afectaba a pequeñas articulaciones de las manos y que cedió en 6 meses con tratamiento antiinflamatorio. En aquella ocasión se objetivó la presencia de anticuerpos antinucleares (ANA) 1/160 patrón homogéneo-moteado. Durante 1995 presentó tres nuevos episodios de dolor y tumefacción oligoarticular (manos, rodillas y tobillos) de corta duración (menos de una semana) y se objetivó la presencia de ANA 1/160 patrón homogéneo-moteado, factor reumatoide 104 U/ml (N < 30), anti-ADN, RNP, Sm, Ro y La negativos. La paciente permaneció asintomática hasta marzo de 1999 cuando inició artritis aditiva en las muñecas, IFP de manos, codos, rodillas y tobillos. En mayo ingresó en el hospital por persistencia de la artritis, a la que se había añadido en los últimos días fiebre de hasta 38 °C, astenia, fenómeno de Raynaud en las manos, las lesiones cutáneas, úlceras orales y edemas maleolares. En la exploración física se objetivó una presión arterial de 100/60 mmHg, frecuencia cardíaca de 75 lat/min, temperatura de 36,8 °C, lesiones cutáneas en forma de pequeñas máculas eritematosas en extremidades superiores e inferiores, así como lesiones papulares en cara dorsal de la tercera metacarpofalángica, una úlcera oral y edema de párpados y maléolos. También se evidenciaron crepitantes en la base izquierda a la auscultación respiratoria, auscultación cardíaca normal, abdomen normal, reflejos y sensibilidad conservada con debilidad 4/5 de los flexores del cuello, deltoides y celda anteroexterna de las piernas, así como tumefacción difusa de los dedos de las manos con artritis de IFP y tumefacción de rodillas. Las exploraciones complementarias evidenciaron: leucocitos, 9.180 (73% N; 15,3% L); hemoglobina, 13,2; plaquetas, 326.000; velocidad de sedimentación globular (VSG), 30; proteína C reactiva (PCR), 12,1 mg/dl (0-0,5); creatinina, 0,9; glucosa, urea, Na-K, fosfatasas alcalinas, GGT, bilirrubina, colesterol y triglicéridos normales. Proteínas totales 62 g/l, GOT/GPT 247/329 U/l (0-31), LDH 2.101 U/l (0-480), CPK 7.044 U/l (0-170), aldolasas 152 U/l (0-7,6). Estudio básico de orina: proteínas ++; sangre +++. Sedimento minutado: hematíes, 0-2 por campo; cilindros, 0; leucocitos, 0. Orina de 24 h: aclaramiento de creatinina, 73,4 ml/min; proteinuria de 1,72 g/24 h, mioglobina 18.265 µg/24 h (0-50). Uriproteinograma compatible con proteinuria fisiológica. Serologías de citomegalovirus, toxoplasma, parvovirus B-19, virus de Epstein-Barr y VIH, negativas. Hemocultivos negativos. ANA 1/320 patrón homogéneo, anticuerpos antimúsculo liso, antimitocondriales, anticélula parietal, anti-ADN, anti-RNP, anti-Sm, anticardiolipina, anti-Ro y anti-La, negativos. C3 75,1 (90-180), C4 20,5 (10-40), CH50 36,3 (35-60). En la radiografía de tórax se evidenciaron pequeñas áreas de opacificación en base izquierda con borramiento del hemidriafragma izquierdo. La gasometría arterial fue normal. La espirometría forzada estuvo dentro de los valores de referencia. Prueba broncodilatadora no significativa. Volúmenes pulmonares: moderada disminución de la capacidad pulmonar. Difusión de CO disminuida, que se corrige con el volumen pulmonar. Tomografía axial computarizada (TAC) torácica: imágenes lineales y pequeñas áreas de panalización en bases pulmonares compatibles con fibrosis. Neurografía sensitiva peroneal: normal. Neurografía motora peroneal: disminución de la amplitud del potencial evocado. Electromiograma: moderada presencia de actividad espontánea con abundantes polifásicos y patrón de reclutamiento interferencial temprano en la región deltoidea y tibial anterior. Electrocardiograma y ecocardiograma: normales.
Se practica una prueba diagnóstica.
Diagnóstico diferencial
Dr. Sergi Ordóñez. En resumen, se trata de una mujer de 27 años que presenta una sintomatología sistémica compleja así como numerosas alteraciones en las exploraciones complementarias realizadas, por lo que es necesario recurrir a un síntoma/signo guía que, además de ser el más significativo, permita realizar un diagnóstico diferencial y establecer así un juicio diagnóstico razonable.
Al leer el caso, se observa cómo la afección muscular desempeña un papel destacado por lo que nos basaremos en este dato para establecer el diagnóstico diferencial. La afección muscular la podemos analizar desde tres puntos de vista diferentes, según los datos aportados:
En primer lugar, destaca la presentación subclínica. La enferma no refiere la presencia de debilidad ni mialgias y es en la exploración física donde se objetiva una leve debilidad de los músculos flexores del cuello, del deltoides y de la musculatura de la celda anteroexterna de las piernas, que permite definir la afección muscular como de tipo proximal y simétrico. Este patrón de afección muscular, junto con una masa muscular conservada y unos reflejos normales, orientan la debilidad hacia un proceso de tipo miopático.
Desde el punto de vista analítico, destaca una importante elevación de todas las enzimas musculares que traducen una destrucción muscular activa y confirman la presencia de una miopatía. Especial importancia adquieren los valores de creatincinasa, ya que son los que presentan una mayor sensibilidad y una mayor correlación con la intensidad de la lesión1.
En tercer lugar, el estudio electromiográfico, en el que se observa una disminución de la amplitud de los potenciales, actividad espontánea, abundantes polifásicos y un patrón interferencial temprano, todas ellas características de un proceso miopático.
Así pues, y una vez descartada una causa neurológica de la debilidad, tanto por la exploración física como por las exploraciones complementarias realizadas, nos encontramos ante una paciente con una miopatía adquirida de tipo proximal y simétrico ante la cual se plantea un amplio diagnóstico diferencial que analizaremos a continuación.
Miopatías tóxicas o por fármacos. La elevación de las creatincinasas y/o el desarrollo de mialgias en un paciente que se halle en tratamiento farmacológico debe hacer considerar la existencia de una miopatía tóxica. Los fibratos, las estatinas, la colchicina, la ciclosporina, la d-penicilamina, la zidovudina, la cloroquina y los glucocorticoides son algunos de los fármacos implicados en este tipo de miopatías2. Dentro de este grupo también se incluirían sustancias como el alcohol y la heroína.
La sintomatología sistémica de la paciente unida a la ausencia de fármacos u otros tóxicos permite descartar esta etiología.
Miopatías endocrinas. Dentro de este grupo se incluyen los trastornos de la glándula tiroides, paratiroides y de las suprarrenales.
Entre todas ellas cabe destacar el hipotiroidismo, que puede provocar una miopatía simétrica similar a la polimiositis (PM) con elevación de las enzimas musculares3,4. Aunque en este caso no se han determinado las hormonas tiroideas, la ausencia de otros síntomas de hipotiroidismo así como la existencia de unos reflejos normales permiten descartar de forma razonable esta etiología.
El hipertiroidismo también puede producir diferentes tipos de afección muscular aunque con enzimas musculares normales.
Miopatías infecciosas. Numerosas infecciones pueden ser causa de miopatía. Dentro del grupo de miopatías de origen viral cabe destacar la afección muscular descrita en pacientes infectados por el VIH, que puede ser indistinguible de una verdadera PM o de una dermatomiositis (DM)5,6. Estos pacientes también pueden desarrollar miopatía asociada al tratamiento con zidovudina (AZT), que remite tras la retirada del fármaco6.
El antecedente de traumatismo muscular en un paciente que presenta fiebre elevada, dolor muscular intenso y leucocitosis con neutrofilia debe hacernos pensar en una miositis de origen bacteriano. Entre ellas cabe destacar la miositis estafilocócica o piomiositis tropical, las infecciones por el estreptococo betahemolítico del grupo A y las causadas por gérmenes anaerobios como el Clostridium sp.
Entre las infecciones parasitarias que pueden causar miositis se encuentra la toxoplasmosis, la tripanosomiasis, la cistercosis y la triquinosis. Las miositis fúngicas, por otro lado, son infrecuentes.
Las características clínicas de la enferma, la ausencia de leucocitosis y la negatividad de los hemocultivos y de las serologías realizadas permiten descartar la etiología infecciosa de la miopatía.
Miopatías inflamatorias idiopáticas (MII). La presencia de una miopatía proximal y simétrica, acompañada o no de lesiones cutáneas o de otros síntomas sistémicos, obliga siempre a descartar una miopatía inflamatoria idiopática tipo polimiositis-dermatomiositis. Si recordamos los criterios diagnósticos de ambas (tabla 1), observaremos cómo la paciente del caso que nos ocupa cumple criterios definidos de DM, dado que presenta tres de los cuatro primeros criterios más el quinto criterio, considerando las lesiones de las terceras metacarpofalángicas como compatibles con pápulas de Gottron.

Dentro del grupo de MII existen otros subgrupos que deben tenerse en cuenta al realizar un diagnóstico diferencial.
1. MII asociadas a otras conectivopatías. Un 20% de las PM/DM presentan otra enfermedad autoinmune asociada como la esclerodermia, el lupus eritematoso sistémico, la artritis reumatoide, la enfermedad mixta del tejido conectivo y el síndrome de Sjögren.
Aunque la debilidad muscular que presentan los pacientes con esclerodermia suele ser secundaria a atrofia por desuso, en un 10-15% de ellos se encuentra evidencia clínica y analítica de miositis. Aunque la enferma presenta algunas características clínicas frecuentes en pacientes con esclerodermia no podemos diagnosticarla al no cumplir criterios de esta enfermedad. Recordar además la existencia del síndrome de superposición miositis-esclerodermia, en el que en el 25% de los casos se encuentran anticuerpos anti PM-Scl.
Los pacientes con lupus eritematoso sistémico (LES) pueden padecer miopatía inflamatoria que cursa con debilidad muscular proximal y elevación enzimática. La paciente cumple cuatro criterios de LES (artritis, úlceras orales, proteinuria > 0,5 g/24 h, y ANA positivo), por lo que de cara al diagnóstico final debe tenerse en cuenta esta entidad.
Aunque la paciente no presenta una artritis reumatoide, es interesante recordar la existencia en estos enfermos de la llamada miositis reumatoidea, que cursa con necrosis fibrilar y elevación de las creatincinasas.
La enfermedad mixta del tejido conectivo (EMTC) podría ser otra entidad a considerar, pero la ausencia de anticuerpos anti-RNP, indispensables tanto en los criterios de clasificación de Alarcón-Segovia como en los de Kasukawa, permite descartar esta entidad.
2. PM/DM asociada a neoplasia. En todo paciente con sospecha de PM/DM debe descartarse enfermedad neoplásica asociada. Aunque no se ha realizado específicamente cribado de neoplasia, el cuadro clínico, la evolución y las exploraciones complementarias realizadas no apoyan esta asociación, siendo además rara su presencia en menores de 40 años.
3. Miositis por cuerpos de inclusión. Afecta con mayor frecuencia a varones de más de 50 años. Se caracteriza por afectar a grupos musculares proximales y distales, con cierto grado de atrofia y sin afección cutánea. Las creatincinasas suelen ser normales o discretamente elevadas.
4. Existen otras formas mucho menos frecuentes dentro del grupo de MII que pueden descartarse clínicamente, como la miositis focal nodular, la miositis eosinofílica, la miositis orbitaria, la miositis de células gigantes y la miositis osificante.
Una vez realizado el diagnóstico diferencial del cuadro miopático y teniendo como posibilidades diagnósticas una MII tipo DM o un LES, sería interesante analizar la afección renal y la pulmonar.
La afección renal en las MII tipo DM/PM es rara2. De forma ocasional se encuentra proteinuria, hematuria microscópica, piuria y síndrome nefrótico. En la biopsia renal podemos hallar desde lesiones glomerulares mínimas hasta glomerulonefritis extramembranosa e incluso depósitos amiloides7. Asimismo debe tenerse en cuenta la presencia de mioglobinuria secundaria a la destrucción muscular.
Al analizar las alteraciones renales, destaca la presencia de mioglobinuria, de una proteinuria moderada y según la tira de orina o perfil de orina reciente, de sangre. La función renal, en cambio, está conservada. Es obligado en todo paciente que presenta sangre en la tira de orina realizar un sedimento minutado, que nos permitirá diferenciar si el pigmento detectado por la tira es realmente sangre (la tira de orina será positiva y el sedimento detectará la presencia de hematíes) o es mioglobina (la tira también será positiva pero en el sedimento no se observarán hematíes). El resultado del sedimento fue de 0-2 hematíes por campo, dato que permite descartar la presencia de hematuria y atribuir la positividad de la tira de orina a la presencia de mioglobina.
Se conoce, en casos de rabdomiólisis, la lesión renal que la mioglobina provoca en forma de necrosis tubular aguda y que cursa con insuficiencia renal. Se considera que son necesarias concentraciones de mioglobina superiores a 0,25 mg/ml para que ésta lesione la vía urinaria y provoque insuficiencia renal8. Aun así existen casos en la bibliografía en que pacientes con cifras de mioglobina muy superiores no desarrollan fracaso renal.
En segundo lugar, destaca la presencia de una proteinuria moderada que no se acompaña de hematuria ni de insuficiencia renal. La proteinuria fisiológica (< 150 mg/día) está compuesta por un 50% de proteínas de origen plasmático, la mayoría de las cuales se reabsorben en los túbulos, y un 50% de proteínas que se originan en los túbulos y en el tracto urinario.
La proteinuria patológica puede tener tres orígenes según el mecanismo implicado:
Proteinuria glomerular (por aumento de la permeabilidad glomerular).
Proteinuria tubular (por disminución de la reabsorción tubular proteica).
Proteinuria por sobrecarga filtrada (por proteínas que debido a su elevada concentración en plasma son filtradas en gran cantidad y sobrepasan la capacidad tubular de reabsorción).
La ausencia de hematuria, hipoproteinemia y albuminuria (el resultado del uriproteinograma indica que no hay predominio de albúmina) permiten descartar una lesión glomerular como causa de la proteinuria. El mecanismo más probable sería el de una proteinuria de origen mixto (tubular/sobrecarga filtrada) donde los valores elevados de mioglobina en sangre atravesarían el glomérulo y provocarían la lesión de los túbulos, afectando así a la reabsorción de las proteínas de bajo peso molecular filtradas. Al mismo tiempo, se afectaría a la reabsorción tubular de mioglobina al sobrepasar ésta la capacidad de reabsorción.
Desde el punto de vista respiratorio las pruebas funcionales señalan la existencia de un patrón restrictivo de afección intersticial, con disminución de la capacidad pulmonar y de la transferencia de CO. En la radiografía de tórax (fig. 1) se observa una discreta elevación del hemidiafragma izquierdo. La presencia de una pequeña atelectasia en la base izquierda podría explicar esta retracción, pero la TAC practicada (fig. 2) permite observar áreas de panalización en ambas bases compatibles con fibrosis, que serían la etiología más probable de dicha retracción.
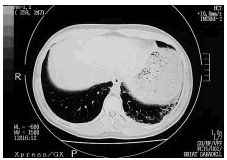
Figura 1. Tomografía axial computarizada (TAC) torácica: áreas de panalización en ambas bases pulmonares.

Figura 2. Tinción con hematoxilina-eosina. Fibras musculares en sección transversal, con cambios degenerativos y regenerativos. Infiltrado linfocitario perivascular y perifascicular.
Ciertas miopatías pueden afectar a la deglución, produciendo microaspiraciones. Éstas, en ocasiones, pueden dar lugar a imágenes similares a las observadas en la TAC pero en la historia clínica no se recoge que la paciente presente disfagia, por lo que la musculatura en esta zona debe considerase indemne. En el contexto de miositis puede afectarse también la musculatura diafragmática dando lugar a una disminución del tamaño pulmonar, por lo que también debe tenerse en cuenta en esta enferma.
En resumen, nos encontramos ante una paciente que presenta miositis, proteinuria mixta no glomerular, fibrosis pulmonar, artritis, Raynaud, úlceras orales, lesiones cutáneas sugestivas de DM, ANA positivo y factor reumatoide positivo.
Anteriormente se ha considerado tanto al LES como a la DM como posibles diagnósticos. En el caso del LES, si de los cuatro criterios diagnósticos que cumplía la paciente se elimina la proteinuria (la proteinuria del LES se debe a glomerulopatía), nos encontramos ante una paciente que cumple únicamente tres criterios. Si a esto se añade la ausencia de leucolinfopenia, anti-ADN o anti-Sm, el diagnóstico de LES se convierte aún en más improbable, por lo que éste podría descartarse de forma razonable.
En 1992 se propuso una clasificación de las miopatías inflamatorias idiopáticas basada en la tipificación del HLA y, sobre todo, en la detección de determinados anticuerpos, lo que permitió definir una serie de subgrupos. Dentro de estos nuevos subgrupos, el más representativo y frecuente es el denominado síndrome antisintetasa, en el que una serie de características clínicas va unida a la presencia de anticuerpos contra las sintetasas citoplasmáticas del ARN de transferencia. Se han descrito 5 anticuerpos antisintetasas asociados a las miopatías inflamatorias, de los cuales el más común es el anti-Jo1 (presente en el 20-40% de enfermos con PM o DM). El resto, con una frecuencia menor al 3%, corresponden al anti-PL-7, anti-PL-12, anti-EJ y anti-OJ. Las principales manifestaciones clínicas de este síndrome junto con su frecuencia de aparición están recogidas en la tabla 29.

Otros anticuerpos unidos a la MII son los anti-SRP y los anti-Mi2. Los anticuerpos anti-SRP, presentes en un 5% de las PM, han sido descritos en mujeres de raza negra con miositis aguda y sin afección cutánea. Los anti-Mi2, presentes en un 20% de las DM y con una especificidad del 95% para esta enfermedad, se caracterizan por una DM clásica con afección en casi el 100% de los casos de la V de cuello.
Teniendo en cuenta todas estas consideraciones: ¿cuál sería la prueba diagnóstica definitiva?
Dado que la enferma cumple criterios de DM y que la biopsia muscular más que una prueba diagnóstica sería una prueba de confirmación, creemos que la determinación de los anticuerpos antisintetasa, principalmente el anti-Jo1 dada su mayor frecuencia, constituiría la prueba diagnóstica definitiva, ya que su positividad permitiría diagnosticar a la enferma de un síndrome antisintetatasa.
Diagnóstico del Dr. Ordóñez
Dermatomiositis. Síndrome antisintetasa.
Discusión
Dra. Marta Larrosa: Tal como ha comentado el Dr. Sergi Ordóñez la prueba diagnóstica que se solicitó fue la determinación de los anticuerpos anti-Jo1 que resultó positiva. Se realizó además una biopsia muscular de región del tibial anterior izquierdo para confirmación de la miositis.
Dra. Isabel Ojanguren: Se recibió un fragmento irregular de tejido muscular de 1,4 * 0,5 cm. La microscopia óptica demuestra algunos fascículos prácticamente normales, y otros con múltiples cambios degenerativos y regenerativos. Estas alteraciones se acompañan de un infiltrado linfocitario perivascular y perifascicular que está compuesto por células B (L26) y células T (CD45RB), las más abundantes en las zonas intrafasciculares (fig. 2). Estos cambios son compatibles con dermatomiositis.
Diagnóstico definitivo
Dermatomiositis. Síndrome antisintetasa.