







Introducción
El síndrome de Sjögren (SS) es una enfermedad autoinmune sistémica que se caracteriza fundamentalmente por la presencia de xeroftalmía y xerostomía1,2, así como la existencia de infiltración linfoplasmocitaria de las glándulas de secreción exocrina. Si bien la sequedad ocular y bucal son los síntomas más frecuentes en el SS, pueden aparecer otras manifestaciones clínicas en el curso evolutivo de la enfermedad. En la gran mayoría de casos, la infiltración linfocitaria queda confinada al tejido glandular salival y lagrimal, pero en ocasiones puede extenderse a localizaciones extraglandulares. El SS es probablemente la enfermedad autoinmune más frecuente en nuestro medio, aunque su habitual pobreza sintomatológica, especialmente en estadios evolutivos tempranos, ocasiona que esté a menudo infradiagnosticada. Afecta predominantemente al sexo femenino, con una relación de 9-10:1 respecto al masculino. La edad de aparición del SS se sitúa habitualmente entre los 40 y los
60 años3, aunque también se han descrito casos en edades más tempranas de la vida.
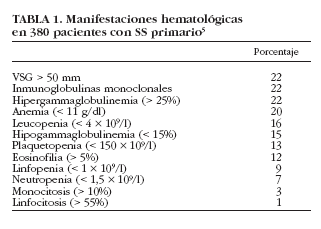
Al igual que en otras enfermedades autoinmunes sistémicas, las alteraciones analíticas son frecuentes en los pacientes con SS e incluso pueden ser la primera manifestación en algunos pacientes. Diversas alteraciones analíticas, como una VSG elevada, la existencia de hipergammaglobulinemia o la presencia de ciertas hemocitopenias (anemia, leucopenia o plaquetopenia) son datos que se pueden detectar habitualmente en los pacientes con SS tanto primario como secundario.
Alteraciones del proteinograma
Las anormalidades en la concentración de proteínas séricas contribuyen, al igual que las alteraciones hematológicas, al gran espectro de manifestaciones biológicas que conforman el SS. Las proteínas totales pueden ser superiores a 75 g/l en un 80% de los casos, y en el proteinograma, las fracciones alfa-1 y alfa-2 pueden estar elevadas en un 20% de los casos4. En nuestra serie de pacientes con SS primario hemos detectado una elevación de las fracciones alfa-1 en un 54%, alfa-2 en un 20% y beta en un 61%5. De todas formas, las principales alteraciones que se observan en el proteinograma de los pacientes con SS se centran en la fracción de las gammaglobulinas.
Hipergammaglobulinemia policlonal
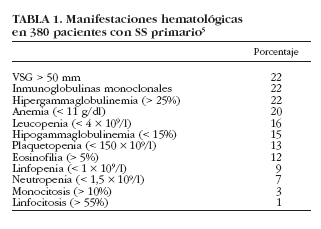
La hipergammaglobulinemia policlonal es uno de los datos analíticos más característicos del SS, ya que refleja la hiperactividad linfocitaria característica de la enfermedad. Aunque se ha descrito tanto en el SS primario como en el secundario, ya en los estudios clásicos de los años setenta se consideró como un dato más representativo de la forma primaria de la enfermedad. En dichos estudios6,7 se describe la existencia de hipergammaglobulinemia en casi el 100% de los pacientes con SS, pero estudios posteriores describen porcentajes inferiores (36-42%)6-10. Alexander et al8 encontraron cifras > 4 g/l en el 36% de pacientes, mientras que en un estudio reciente, Skopouli et al10 describen hipergammaglobulinemia (> 2 g/l) en el 42% de sus pacientes; en nuestra serie hemos detectado una hipergammaglobulinemia (> 25% de las proteínas totales) en un 22% de casos5 (tabla 1).
Los trastornos clínicos debidos a la elevación de las gammaglobulinas son excepcionales. No suelen observarse fenómenos de hiperviscosidad ni defectos en la acidificación renal causados por la hipergammaglobulinemia, aunque Skopouli et al10 describieron el caso de un paciente con una importante
hipergammaglobulinemia que murió por un tromboembolismo pulmonar. En cambio, la relación entre hipergammaglobulinemia y púrpura cutánea es más conocida y fue descrita en pacientes con SS en los años sesenta y setenta bajo el nombre de púrpura hipergammaglobulinémica de Waldenstrom, considerándose incluso como una entidad independiente11,12. Posteriormente, se ha comprobado que la existencia de púrpura e hipergammaglobulinemia tal vez forme parte del conjunto de manifestaciones extraglandulares y biológicas asociadas al SS. Finalmente, la existencia de hipergammaglobulinemia se ha relacionado con la positividad de los anticuerpos anti-Ro/SS-A. En nuestros pacientes con hipergammaglobulinemia hemos observado una mayor prevalencia de manifestaciones extraglandulares (adenopatías, vasculitis cutánea, neuropatía periférica, fenómeno de Raynaud), pruebas diagnósticas positivas (gammagrafía parotídea, biopsia de glándula salival) y presencia de autoanticuerpos (ANA, anti-Ro/SS-A, anti-La/SS-B, y FR)5. Además, los valores de gammaglobulinas se correlacionaban de forma estadísticamente significativa con los valores de VSG, hemoglobina y recuento leucocitario5. Otros estudios han descrito resultados parecidos. Davidson et al13 encontraron una mayor concentración de IgG sérica en pacientes Ro/La+, y una VSG más elevada en aquellos pacientes con ANA/FR+ y Ro/La+. Asimismo, Markusse et al14 reportaron que la presencia de autoanticuerpos anti-Ro/SS-A y anti-La/SS-B se correlacionaba con la existencia de hipergammaglobulinemia. Por tanto, parece existir una estrecha relación entre hipergammaglobulinemia, VSG y anticuerpos positivos en el SS.
Hipogammaglobulinemia
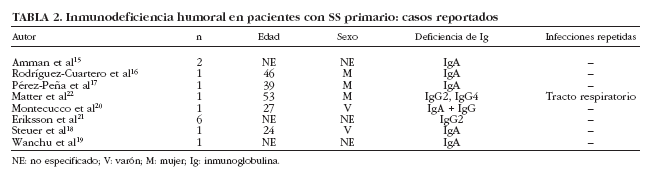
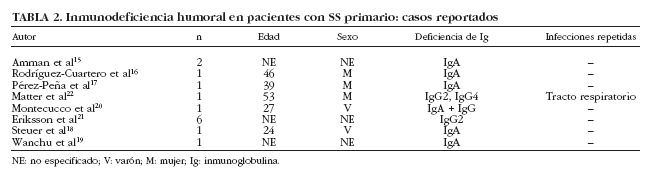
La presencia de hipogammaglobulinemia en pacientes con SS se ha considerado clásicamente como un dato sugestivo de linfoproliferación. Bloch et al6 encontraron una prevalencia del 3% (2 de 62 pacientes) y ambos pacientes presentaron reticulosarcomas. En nuestra serie, hemos detectado que un 20% (76 de 380) de pacientes con SS presentan cifras bajas (< 1,5 g/l) de gammaglobulinas5. En nuestros pacientes con hipogammaglobulinemia, tres características específicas merecen especial atención. La primera es que 4 de los 8 pacientes con linfoma presentaban hipogammaglobulinemia, confirmando así su asociación con el desarrollo de enfermedades linfoproliferativas. En segundo lugar, nuestros pacientes con hipogammaglobulinemia presentaron un muy bajo porcentaje de anticuerpos positivos como ANA (53%), anti-Ro/SS-A (11%) y anti-La/SS-B (3%), lo que podría sugerir una respuesta inmunológica distinta de este subgrupo de pacientes o bien la existencia de resultados falsos negativos debido a los valores reducidos de inmunoglobulinas circulantes. Finalmente, identificamos en nuestra serie de pacientes con SS primario, 3 casos de inmunodeficiencia humoral, 2 casos de inmunodeficiencia variable común (IVC) y un caso de deficiencia selectiva de IgA. La inmunodeficiencia descrita con más frecuencia en el SS es el déficit de IgA, aunque debe considerarse como una asociación muy infrecuente ya que desde 1971 sólo se han reportado 7 casos20 (tabla 2). De los 7 pacientes descritos con SS y déficit selectivo de IgA, sólo se describen infecciones de repetición en un paciente y episodios de parotiditis en otros 2 pacientes. Con todos estos datos, consideramos que debe realizarse la cuantificación de los subtipos de Ig sólo en aquellos pacientes con SS con sospecha de inmunodeficiencias humorales (p. ej., aquellos con infecciones repetidas) y no debería considerarse como una prueba de laboratorio rutinaria a realizar en todo paciente con SS.
Alteraciones de las concentraciones de las distintas inmunoglobulinas
Respecto a la elevación específica de los subtipos de inmunoglobulinas, lo más frecuente es el aumento de la IgG (> 1,5 g/l), que se ha reportado en 80% de los casos4. En algún estudio se ha demostrado que los pacientes con síndrome seco y mayores concentraciones de IgG acaban por desarrollar con más frecuencia un SS primario23. Los valores de IgA suelen ser normales, aunque los de IgA secretoria pueden ser el doble de lo normal4. Si bien los valores de IgM generalmente son normales4, se han observado elevaciones policlonales en pacientes con SS asociado a esclerodermia o a la presencia de valores elevados de factor reumatoide. En nuestra serie de pacientes con SS primario, el análisis de los tipos de inmunoglobulinas en los pacientes con hipogammaglobulinemia demostró la existencia de títulos bajos de IgG en 10, de IgA en 5 y de IgM en 3 pacientes5.
Otro trastorno menos frecuente es el déficit selectivo de IgG2, que se ha relacionado con una mayor susceptibilidad a infecciones bacterianas neumocócicas22. Eriksson et al21 describen que aunque los pacientes con SS puedan tener un déficit de IgG2 a pesar de tener valores elevados de IgG, éste no predispone por sí solo a infecciones.
Reactantes séricos de fase aguda
Velocidad de sedimentación
La velocidad de sedimentación globular (VSG) suele estar frecuentemente elevada en los pacientes con SS. En muchas ocasiones, los pacientes con VSG elevada presentan, a su vez, cifras aumentadas de las proteínas totales plasmáticas. Además, la existencia de VSG elevada se ha descrito como un factor predictivo de evolución a SS en pacientes con síndrome seco que inicialmente no cumplían criterios para SS23.
En nuestra serie hemos detectado una VSG > 50 mm/h en 82 (22%) pacientes, siendo > 100 mm/h en 15 (4%)5. Cuando se compararon con pacientes con VSG < 50 mm/h, aquellos con VSG > 50 mm/h presentaron una mayor prevalencia de tiroiditis, afección renal, vasculitis cutánea, neuropatía periférica, gammagrafía parotídea positiva, biopsia salival positiva, anti-La/SS-B, FR, crioglobulinemia e hipocomplementemia. Además, los valores de VSG se correlacionaron con los de hemoglobina. Encontramos también una relación estrecha entre los valores de VSG y los porcentajes de gammaglobulinas, lo que sugiere que la existencia de VSG elevada en un paciente con SS puede estar directamente relacionada con la existencia de valores elevados de gammaglobulinas circulantes. La VSG podría ser un marcador biológico útil en el SS primario para identificar aquellos pacientes con una mayor hiperactividad policlonal de células B, ya que está relacionada no sólo con la presencia de hipergammaglobulinemia y FR, sino también con una mayor frecuencia de autoanticuerpos circulantes como ANA, anti-Ro/SS-A y anti-La/SS-B.
Proteína C reactiva
Aunque no se han realizado estudios sobre el papel diagnóstico de la proteína C reactiva (PCR) en el SS, en un estudio preliminar24 se han descrito valores normales en 22 de 24 pacientes con SS primario respecto a aquellos con SS asociado a artritis reumatoide. Por otra parte, Davidsson et al13 no encontraron diferencias significativas respecto al valor medio de la PCR en pacientes con SS primario agrupados según su perfil inmunológico (seronegativo, ANA/FR o Ro/La). Es posible que la principal utilidad de la PCR en el SS primario pueda ser el diagnóstico diferencial entre infección y actividad sistémica de la enfermedad.
ß2 microglobulina
La ß2 microglobulina es una proteína de bajo peso molecular (11.700 daltons), secretada por diversas células nucleadas, que normalmente se encuentra en concentraciones bajas en suero, líquidos corporales y secreciones. Los valores de ß2 microglobulina en suero se encuentran aumentados en pacientes con SS25, un dato analítico considerado como un factor predictivo de evolución a SS primario en pacientes con síndrome seco23. Por otra parte, se han detectado valores elevados de ß2 microglobulina en las glándulas salivales de pacientes con SS primario, lo que sugiere una posible relación con la infiltración linfocitaria. En un estudio realizado en pacientes con SS primario se encontró una concentración elevada en saliva de la ß2 microglobulina (> 4 µg/ml) en un 35% de casos25.
Además de la elevación de los niveles de ß2 microglobulina en glándulas salivales, también se han encontrado valores significativos de esta proteína en suero de pacientes con SS y diversas afecciones extraglandulares, especialmente pulmonares, nefrológicas y linfoproliferativas. Se han detectado concentraciones elevadas de la ß2 microglobulina en pacientes con enfisema pulmonar26 o alveolitis linfocítica/neutrofílica27. También se han observado valores elevados de ß2 microglobulina en suero de pacientes con acidosis tubular renal respecto a los pacientes con capacidad de acidificación normal28. Finalmente, se ha relacionado la existencia de concentraciones elevadas de la ß2 microglobulina con la presencia de tumores malignos. Michalsky et al25 encontraron elevada la ß2 microglobulina en el 77% de pacientes con SS asociado a linfoma o seudolinfoma, aunque no existen estudios posteriores que confirmen estos datos.
De acuerdo con lo descrito anteriormente, la medición de la ß2 microglobulina en saliva y suero podría ser un método útil y sencillo para estimar el grado de infiltración linfocitaria activa en los órganos afectados por el SS. También se podría utilizar como un parámetro de laboratorio en el seguimiento clínico del paciente, a fin de descartar una posible evolución a linfoproliferación, ya que el incremento en la proliferación y el aumento de masa linfoidea se correlaciona con los valores de ß2 microglobulina.
Hemocitopenias
Anemia
La anemia está presente, según las distintas series, entre el 16 y el 50% de los pacientes con SS6-8,10,29 (tabla 3). En general, el tipo más común de anemia es el mismo que se observa en las enfermedades inflamatorias crónicas, es decir, normocítica y normocrómica4,7. No es frecuente identificar una causa específica de la anemia en el paciente con SS. En 1965, Bloch et al6 describieron 2 casos de pacientes con una anemia ferropénica y otro con un síndrome de Felty asociado. Ramakrishna et al29 encontraron anemia en 10 de 27 pacientes (37%), aunque sólo describieron una causa evidente de anemia en 7 de ellos (anemia hemolítica en 3, anemia refractaria asociada a síndrome mielodisplásico en dos, aplasia pura de la serie eritrocitaria en uno y anemia aplásica en otro). Finalmente, en nuestra serie de pacientes5 hemos descrito 76 con anemia (Hb< 11 g/l), aunque sólo 15 (4%) presentaron una anemia grave (Hb < 9 g/l); 71 pacientes (93%) presentaron una anemia normocítica normocrómica, 3 (4%) una anemia microcítica y 2 (3%) anemia macrocítica. En 21 pacientes se pudo identificar una causa específica de la anemia, como hemorragia digestiva, gastritis atrófica crónica, enfermedades linfoproliferativas, anemia hemolítica, síndrome mielodisplásico y anemia aplásica. En los restantes 55 pacientes, no se identificó ninguna causa específica, aunque no pudimos excluir una hemólisis subclínica (no se realizó la prueba de Coombs ya que ninguno presentaba evidencia de hemólisis biológica). Finalmente, y al efectuar una comparación con los pacientes con valores normales de hemoglobina, aquellos con anemia presentaron una prevalencia mayor de afección renal, vasculitis cutánea, neuropatía periférica, ANA, FR, crioglobulinemia e hipocomplementemia5.

Anemia hemolítica. Aunque algunos estudios demuestran que puede observarse una prueba de Coombs positiva en un 22-47% de pacientes con SS, la hemólisis franca en el SS es poco común30,31, ya que hasta la fecha se han descrito sólo 22 pacientes con SS primario y anemia hemolítica autoinmune (AHAI)8,10,20,24,30,32-35,38 (tabla 4). La AHAI se ha descrito también en pacientes con SS secundario (asociado a artritis reumatoide, lupus eritematoso sistémico o esclerodermia) o asociado a procesos linfoproliferativos malignos32. De forma excepcional, la AHAI puede ser la primera manifestación hematológica de un SS incipiente. De los casos con SS y AHAI publicados, la mayoría se trató con corticoides, presentando una buena respuesta, aunque en 3 pacientes fue necesario añadir un tratamiento inmunosupresor (azatioprina en 2 casos y ciclofosfamida en uno)20,29,30,32,33,35 (tabla 5). Generalmente, estos pacientes presentaron una sintomatología relacionada con anemia aguda (debilidad, palidez, disnea) y una disminución significativa de los valores de hemoglobina.


La causa de la anemia hemolítica en el SS es desconocida, aunque debido a la hiperactividad linfocitaria típica de la enfermedad no se puede descartar la existencia de anticuerpos antieritrocito como causantes de la hemólisis32. Además, cabe resaltar la relación que se ha encontrado entre la anemia hemolítica y la expresión anómala de antígenos Ro y La en la membrana celular, provocada por la intervención de factores exógenos como la exposición a rayos ultravioleta o la infección por virus con un determinado tropismo medular, que provocarían la lisis por autoanticuerpos de los hematíes29. Recientemente, hemos descrito en pacientes con SS la asociación de hemocitopenia con la evidencia de infección por parvovirus B1936, un virus con un marcado tropismo por la médula ósea37. También se ha descrito un caso de anemia hemolítica en un recién nacido de una madre con SS portadora de anticuerpo anti-Ro/SSA, lo que apoyaría la implicación etiopatogénica de dichos autoanticuerpos38.
Desde el punto de vista de la práctica clínica y debido a la escasa prevalencia de la anemia hemolítica en pacientes con SS primario, consideramos que las pruebas hemolíticas (haptoglobina, prueba de Coombs) deberían realizarse sólo en aquellos pacientes con evidencia clínica de anemia aguda y/o evidencia biológica de hemólisis (valores elevados de LDH y bilirrubina).
Anemia perniciosa. Es rara en el SS, aunque puede ser la consecuencia final de una gastritis atrófica crónica, que es la afección gastrointestinal más común39. En 1950 se publicó el primer caso de anemia perniciosa en un paciente con SS40 y en 1970 se describieron dos casos de síndrome seco en una amplia serie de pacientes con anemia perniciosa41. Recientemente, Pedro-Botet et al39 han descrito un caso adicional. Es probable que la inflamación crónica gástrica conduzca a atrofia y consecuentemente a anemia perniciosa. Sin embargo, la gastritis atrófica crónica no es suficiente para el desarrollo de anemia perniciosa, debido al patrón parcheado de la gastritis atrófica crónica con zonas libres de afección. Algunos estudios sugieren que la gastritis atrófica crónica en el SS está causada por la propia enfermedad de base, pues se han encontrado infiltrados mononucleares en la mucosa gástrica39. Por otra parte, aunque los anticuerpos anticélula parietal podrían tener un significado diagnóstico en la enfermedad, su frecuencia en el SS varía ampliamente, entre el 10 al 50%, y no se ha demostrado una clara asociación con manifestaciones clínicas42.
Aplasia medular. Aunque el primer caso de anemia aplásica en el SS se describió en un paciente con linfoma, se han publicado 4 casos adicionales no asociados a procesos linfoproliferativos29,43-45 (tabla 4). Hasta la fecha, no se han realizado estudios etiopatogénicos en pacientes con SS y anemia aplásica. Es probable que exista un mecanismo autoinmune similar al descrito en el lupus eritematoso sistémico, en el que se ha demostrado la existencia de anticuerpos IgG (dependientes o no de complemento) que inhiben la proliferación de la médula ósea24.
La aplasia pura de la serie roja se ha descrito de forma excepcional en el SS29,46 (tabla 4), aunque existen casos adicionales en otras enfermedades autoinmunes asociadas como el lupus eritematoso sistémico y la artritis reumatoide. Los pacientes suelen presentar una anemia normocítica y normocrómica grave, asociada a reticulopenia y ausencia de eritroblastos en una médula ósea normal46. Debido a que la aplasia de la serie eritrocitaria puede ser la primera manifestación de una leucemia (linfocítica o linfoblástica) o de un linfoma (de Hodgkin o no hodgkiniano), es importante realizar un seguimiento clínico estricto de los pacientes con SS y aplasia pura46.
Mielodisplasia. Se han descrito algunos casos de síndrome mielodisplásico en pacientes con SS. Ramakrishna et al29 describen 2 casos de pacientes ancianos con síndrome mielodisplásico y anemia refractaria con anillos sideroblásticos, aunque la presencia del síndrome mielodisplásico podría ser un hecho casual no relacionado con el SS y sí con su edad avanzada. El síndrome mielodisplásico puede acompañarse de diversas anormalidades inmunológicas, incluyendo la presencia de anticuerpos antinucleares, aunque no hay estudios que relacionen este hecho con la asociación de enfermedades autoinmunes.
Leucopenia
Entre un 12 y un 33%4 de los pacientes con SS pueden presentar leucopenia6-8,10,47 (tabla 3). Bloch et al7 detectan una prevalencia del 32% (20 de 62 pacientes), la mayoría con cifras cercanas a 3.000/µl, aunque en 8 pacientes oscilaba entre 1.500 y 2.400/µl. Por su parte, Alexander et al8 encontraron 17 pacientes de 75 (23%) con leucopenia, de los cuales 14 mostraban anti-Ro/SS-A positivo, y Aoki et al47 describen una prevalencia similar (26%), mientras que Skopouli et al10, en un estudio con 261 pacientes, encontraron un 12% de ellos con leucopenia. Cabe destacar que se ha descrito la asociación entre la leucopenia y el síndrome de Felty en pacientes con SS asociado a artritis reumatoide4. En nuestra serie, 59 (16%) pacientes presentaban leucopenia < 4 * 109/l, aunque sólo uno presentó una leucopenia grave (< 1 * 109/l). Cuando se compararon los pacientes con SS según la presencia o no de leucopenia, aquellos que la manifestaron tuvieron una mayor prevalencia de neuropatía periférica, FR, crioglobulinemia e hipocomplementemia5.
Linfopenia
Se ha descrito la existencia tanto de linfocitosis como de linfopenia en pacientes con SS. La presencia de linfocitosis en el SS puede observarse en un 10-40% de los pacientes4; por ejemplo, Bloch et al6 encontraron linfocitosis en 7 pacientes de 62 (11,3%). Por otro lado, Aoki et al47 detectaron linfopenia en 35 de 99 pacientes (35,3%) que presentaban menos frecuencia de artralgias, pero mayor prevalencia de anti-Ro/SS-A. Otros autores han reportado la presencia de una linfopenia absoluta en pacientes con SS, principalmente asociado a una artritis reumatoide. La causa de la linfopenia en el SS no se conoce. Henriksson et al48, en un estudio realizado en 214 pacientes con SS, fueron los primeros en demostrar la presencia de anticuerpos anti-CD4 en un 12% de los mismos, aunque no se encontró correlación alguna entre la presencia de anticuerpos anti-CD4 y linfopenia. En nuestra serie se encontró linfopenia en 23 (9%) pacientes. Los sujetos con linfopenia presentaron una mayor prevalencia de afección renal y anti-La/SS-B respecto a aquellos con SS sin linfopenia5.
Neutropenia
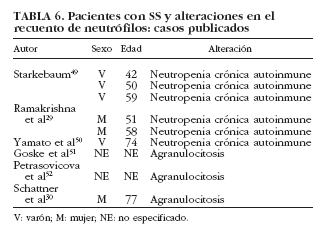
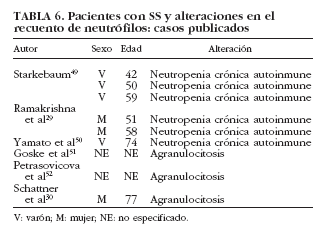
La neutropenia es una manifestación hematológica poco común dentro del SS, siendo más frecuente en enfermedades como la artritis reumatoide y el lupus eritematoso sistémico. Se han publicado 6 casos en pacientes con SS29,30,49,50-52 (tabla 6). De manera puntual se ha correlacionado con la existencia de anticuerpos antineutrófilo50, aunque en la mayoría de los estudios no es posible establecer con claridad el papel de los anticuerpos antipolimorfonucleares en el SS53-55. Finalmente, la presencia de granulocitopenia inmune grave o agranulocitosis es rara en las enfermedades autoinmunes, en general, y en el SS sólo se han reportado 3 casos en la bibliografía30,51,52 (tabla 6).
Plaquetopenia
La alteración plaquetaria en el SS se manifiesta habitualmente como una trombocitopenia leve, con una prevalencia que oscila entre el 8 y el 15% según los estudios6,8,29,47 (tabla 3). Bloch et al6 encontraron trombocitopenia leve (100-150.000/µl) en 5 de 62 pacientes (8%), pero ningún caso de trombocitopenia grave (< 50.000/µl). Ramakrishna et al29, en un estudio realizado con 27 pacientes, describen 5 con casos con trombocitopenia (19%), 4 de ellos diagnosticados de púrpura trombocitopénica idiopática y dos con una buena respuesta al tratamiento con corticoides. Una revisión de la bibliografía japonesa56 reporta 20 pacientes con SS y trombocitopenia, 16 con SS primario y 4 con SS secundario a artritis reumatoide, esclerosis sistémica o polimiositis.
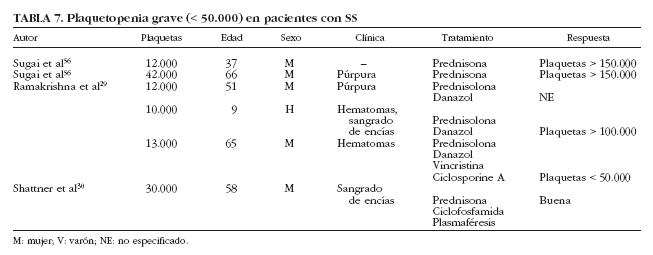
Se han descrito sólo 6 casos de plaquetopenia grave (< 50.000/µl) en pacientes con SS29,30,56 (tabla 7), en su mayoría mujeres entre 37 y 66 años de edad, que presentaban manifestaciones clínicas de trombocitopenia en forma de hematomas, púrpura y sangrado de encías; 4 pacientes fueron tratados con corticoides, además de ciclofosfamida y plasmaféresis en la única paciente que presentó diátesis hemorrágica grave. Por otra parte, se ha descrito un caso de trombocitopenia grave en pacientes con SS de causa farmacológica. Haro et al57 describen el caso de un paciente con SS que desarrolló trombocitopenia grave tras la administración de digoxina. La coexistencia de un SS con otros procesos plaquetopénicos, como la púrpura trombótica trombocitopénica, debe considerarse como un hecho excepcional.
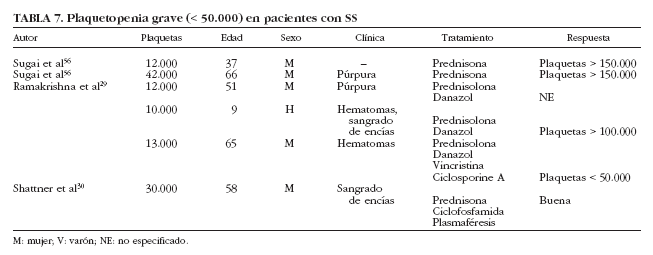
La trombocitopenia en pacientes con SS puede estar causada por la destrucción plaquetaria periférica, ya sea debida a anticuerpos antiplaquetarios o por mediación de inmunocomplejos, de forma similar a lo que se observa en pacientes con LES56. Además, la plaquetopenia se ha relacionado con la presencia de anticuerpos anti-Ro/SS-A y/o anticuerpos anti-La/SS-B. Alexander et al8 encontraron trombocitopenia en un 11% de sus pacientes (8 de 75) con SS, 7 de ellos con anti-Ro (SS-A), y Aoki et al47 encontraron trombocitopenia en 7 de sus 99 (7%), todos con anticuerpos anti-La/SS-B. Es de importancia mencionar que esta anormalidad hematológica fue más frecuente en pacientes jóvenes y varones que en mujeres, los cuales tuvieron una mayor tendencia a presentar afección cutánea, anticuerpos antinucleares y factor reumatoide. En nuestra serie hemos detectado plaquetopenia en 48 (13%) pacientes, aunque sólo 3 presentaron trombocitopenia grave (< 50 * 109/l). Cuando se compararon los pacientes con SS según la presencia o no de trombocitopenia, los primeros presentaron una mayor prevalencia de afección renal y anti-La/SS-B5.
Finalmente, debemos resaltar que la trombocitopenia, al igual que cualquier otra anormalidad hematológica, puede constituir el primer signo de un SS latente30, ya sea primario o incluso secundario a una infección crónica por el VHC58.
Linfoproliferación y alteraciones analíticas
Gammapatía monoclonal
La existencia de una gammapatía monoclonal es un dato analítico frecuente en pacientes con SS. Aunque la aparición de un pico monoclonal cuantitativamente importante puede ser la primera manifestación biológica del desarrollo de un proceso lin-foproliferativo, no es infrecuente observar la existencia de una gammapatía monoclonal de significado incierto (GMSI) no asociada a linfoma en pacientes con SS. Mediante electroforesis de gel de agarosa de alta resolución, capaz de detectar cantidades mínimas de gammaglobulinas indetectables por métodos electroforéticos de rutina, se puede observar cadenas monoclonales en suero en el 47% de los pacientes y en el 76% en orina4. Talal et al describieron la presencia de cadenas ligeras monoclonales en el 100% de los pacientes SS y con manifestaciones extraglandulares frente al 22% de pacientes con síndrome seco59. En un estudio japonés60 se describe una prevalencia de cadenas monoclonales ligeras en el 70% de los pacientes con SS y manifestaciones extraglandulares. En este estudio se describe a 18 pacientes con gammapatía monoclonal 8 de tipo IgA, 4 IgG, 4 IgM y 2 pacientes con una doble banda monoclonal (IgMK/IgGK e IgAK/IgGK).
En nuestra serie de pacientes con SS primario5 detectamos inmunoglobulinas monoclonales en 24 de 110 (22%) pacientes. Comparados con los pacientes con SS con inmunoelectroforesis (IE) negativa, aquellos con una IE positiva presentaron una mayor prevalencia de afección pulmonar y crioglobulinemia. Solamente uno (5%) de estos 22 pacientes con gammapatía monoclonal había desarrollado un linfoma hasta la fecha del estudio. Por tanto, la gammapatía monoclonal es un dato hematológico frecuente, pero no exclusivamente relacionado con la presencia de una neoplasia linfoproliferativa subyacente, ya que en nuestro estudio se ha relacionado con otras manifestaciones, como enfermedad pulmonar o crioglobulinemia. Recomendamos realizar inmunoelectroforesis sérica en todos los pacientes con SS primario por dos razones primordiales. En primer lugar, la detección de una inmunoglobulina monoclonal circulante podría indicar la existencia de una crioglobulinemia asociada que, en algunos casos, podría ocasionar síntomas crioglobulinémicos. En segundo lugar, la presencia de una gammapatía monoclonal y/o crioglobulinemia se asocia con un riesgo mayor de desarrollar enfermedades linfoproliferativas en pacientes con SS primario, como se demuestra en el estudio prospectivo de Tzioufas et al61.
Crioglobulinas
Las crioglobulinas son inmunoglobulinas circulantes que precipitan in vitro con el frío, compuestas por una IgM con actividad FR e IgG policlonal. Desde la primera publicación de crioglobulinemia en un paciente con SS primario62, se han realizado varios estudios que analizan la prevalencia y el significado clínico de las crioglobulinas en los pacientes con SSP61-69. Se ha descrito una prevalencia que oscila entre el 5 y el 61% según las distintas series. Nuestro grupo70 ha analizado la presencia de crioglobulinas en 115 pacientes con SS primario, encontrando una prevalencia del 16%, muy similar a la obtenida por Tzioufas et al61 (19% en 103 pacientes), aunque en las series más limitadas (inferiores a 30 pacientes) la prevalencia había sido superior63,66-68. En nuestros pacientes, el análisis del crioprecipitado demostró un predominio de la crioglobulinemia mixta (CM) tipo II (IgG policlonal + IgM k monoclonal), de acuerdo con la mayoría de los estudios europeos. En países orientales predominan los componentes monoclonales IgA e IgG, algo poco habitual en nuestro medio (tan sólo uno de nuestros pacientes). Finalmente, en uno de los pacientes de nuestra serie se observó una IgG k monoclonal, descrita previamente de manera aislada en algún trabajo72. Al analizar las características clínicas de nuestros pacientes con crioglobulinemia encontramos una mayor prevalencia de vasculitis cutánea, hipocomplementemia e infección asociada por el VHC respecto a los pacientes sin CM. Otros trabajos previos habían encontrado una asociación entre crioglobulinemia y manifestaciones extraglandulares61,63. Tzioufas et al61, en un estudio prospectivo de 103 pacientes con SS primario, han relacionado la presencia de CM monoclonal y los idiotipos CRI 17-109 y G6 presentes en el FR monoclonal con el desarrollo de linfoma, lo cual demuestra que la existencia de CM es un factor predictivo de desarrollo de procesos linfoproliferativos.
En resumen, podemos destacar el papel de las crioglobulinas en el SS en tres importantes aspectos. En primer lugar, una estrecha relación con manifestaciones clínicas de tipo vasculítico. En segundo lugar, su asociación con el VHC, lo que obliga a descartar dicha infección en todo paciente con SS y crioglobulinemia. Finalmente, la presencia de crioglobulinas en un paciente con SS obliga a un mayor control clínico del mismo, debido al mayor riesgo que presenta de evolucionar hacia un proceso linfoproliferativo.
Alteraciones analíticas en el linfoma asociado al síndrome de Sjögren
Una de las principales características de los procesos linfoproliferativos en el paciente con SS es la gran diversidad en su presentación clínica. La mayoría de los linfomas no hodgkinianos (LNH) se presentan con afección del estado general, hepatosplenomegalia y linfadenopatía generalizada, lo cual suele estar asociado con estados avanzados de la enfermedad. En un estudio multicéntrico europeo publicado recientemente73, la mayoría de los pacientes con SS y linfoma presentaron parotidomegalia, fiebre y linfadenopatía, además de una mayor frecuencia de ciertas manifestaciones extraglandulares como vasculitis cutánea y afección del sistema nervioso. En la analítica destacaba una alta frecuencia de citopenias (anemia y leucopenia) y la presencia de inmunoglobulinas monoclonales o crioglobulinas en suero, mientras que la hipogammaglobulinemia fue muy poco frecuente (menos del 10%).
Una de las características más destacadas de los linfomas asociados al SS es la gran frecuencia de afección extranodal, apareciendo la linfoproliferación en determinadas localizaciones. Mediante la revisión de la bibliografía hemos podido constatar la predilección por ciertos órganos sólidos: la mayoría de los linfomas publicados en pacientes con SS se localizan en las glándulas salivales (95 casos), pulmón (23 casos) y estómago (20 casos) [tabla 8]10,13,61,73-79,80-123. La posible aparición de linfoma en cualquier lugar del organismo donde exista tejido linfoide origina una gran variedad de cuadros clínicos de presentación en el paciente con SS como insuficiencia renal hiperuricémica con hipercalcemia, clínica neurológica (habitualmente en linfomas de alto grado), masas renales, testiculares u ováricas. En nuestra serie de pacientes con SS primario5, 7 (3%) desarrollaron enfermedades linfoproliferativas (5 LNH y 2 linfomas de Hodgkin). Las principales anormalidades hematológicas fueron hipogammaglobulinemia (n = 4), linfopenia (n = 3), VSG > 50 mm/h (n = 3), anemia (n = 2), trombocitopenia (n = 2) y leucopenia (n = 2). El perfil inmunológico demostró ANA en 5 de 6 (83%), crioglobulinemia en 2 de 4 (50%), hipocomplementemia en 2 de 6 (33%), anti-Ro/SS-A en uno de 6 (17%) y FR en uno de 6 (17%).

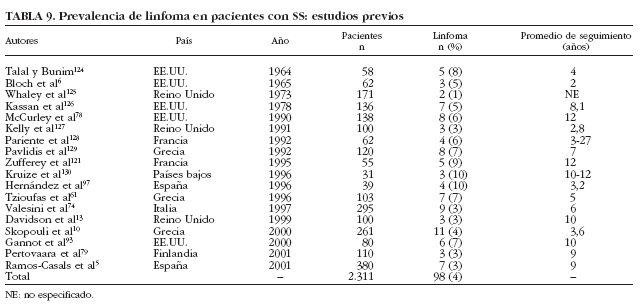
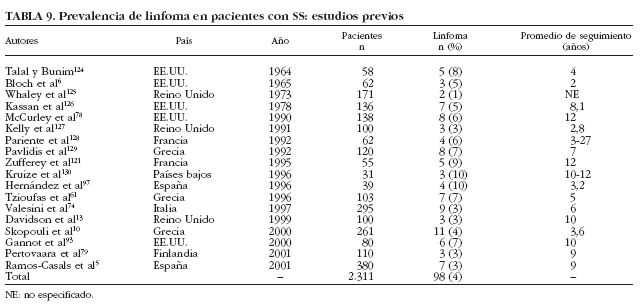
Aunque el linfoma es clásicamente considerado la complicación principal en la historia natural del SS, los estudios transversales han reportado que sólo 98 (4%) de los 2.311 pacientes estudiados con SS primario desarrollaron linfoma6,10,13,61,74,78,79,93,97,121,124-130 (tabla 9). Sólo un trabajo61 ha analizado prospectivamente la incidencia de linfoma, en pacientes con SS primario, describiendo que 7 (7%) de 103 pacientes con esta afección desarrollaron linfoma tras un seguimiento prospectivo de 5 años.
Algunos autores han sugerido que la existencia de determinadas alteraciones analíticas podrían asociarse con un mayor riesgo de desarrollo de linfoma. En 1971, Cummings et al131 describieron una reducción importante de la hipergammaglobulinemia observada justo antes de la aparición del linfoma en pacientes con SS. Otros autores han descrito la disminución en los valores de IgM sérica o negativización del FR132, datos que no se han confirmado en posteriores estudios. Algunos autores describen valores elevados de ß2 microglobulina25 y del receptor soluble de la IL-2133 como marcadores de posible evolución a linfoma. Por último, Tzioufas et al61 han demostrado recientemente que la presencia de crioglobulinemia mixta y de ciertos idiotipos en el FR monoclonal (17-109 y G6) se asocia con un riesgo mayor de desarrollar un linfoma. En una serie de 103 pacientes con SS seguidos prospectivamente durante 5 años, 7 desarrollaron linfoma, y 6 de ellos (83%) tenían crioglobulinas al inicio del estudio frente a 12 (12%) de los 96 que al final no desarrollaron linfoma. Estos datos demuestran que la determinación de crioglobulinas, un dato de laboratorio simple y relativamente fácil de realizar, puede utilizarse como un factor predictivo del posible desarrollo de linfoma en pacientes con SS.
En pacientes con SS y linfoma se han descrito diversas alteraciones cromosómicas, principalmente translocaciones t14,18 o t11,14; también se han descrito otras anomalías como la trisomía del cromosoma 3, mutaciones en el gen p53 o la acumulación del O6-metilguanina-ADN en los linfocitos134. Por otra parte, la aparición de una linfoproliferación es la complicación más grave que pueda surgir a lo largo de la evolución del SS. Además de los factores clínicos predictivos ya conocidos (parotidomegalia persistente, fiebre, adenopatías, esplenomegalia), se ha demostrado de forma prospectiva la importancia predictiva de la aparición en suero de inmunoglobulinas monoclonales o de crioglobulinas y de la detección (mediante técnicas de biología molecular) de una población monoclonal B en la glándula salival. Desde un punto de vista etiopatogénico, la transformación maligna suele ser un proceso de varias etapas que afecta a los linfocitos B, cuya activación policlonal inicial puede desembocar en la selección de alguna clona que posteriormente proliferaría fuera de control134.
Conclusiones
El SS primario demuestra una gran variedad de alteraciones en los parámetros analíticos, incluyendo hemocitopenias, hipergammaglobulinemia o gammapatías monoclonales. Aunque la importancia clínica de estas alteraciones es generalmente leve, algunos pacientes pueden presentar hemocitopenias sintomáticas graves o enfermedades linfoproliferativas malignas. La prevalencia de hemocitopenias, así como la variedad de anormalidades hematológicas, es muy similar a la reportada en pacientes con LES, y aunque la existencia de estas alteraciones hematológicas sí se incluyen en los criterios diagnósticos del LES, no ocurre lo mismo en los criterios diagnósticos de SS. Por ejemplo, en nuestra serie de pacientes con SS primario hemos detectado la existencia de hemocitopenia en 125 (33%), siendo la más frecuente la anemia en 76 (20%), seguida de la leucopenia en 59 (16%) y trombocitopenia en 48 (13%). La mayoría de los estudios han descrito la asociación de leucopenia, linfopenia, anemia y plaquetopenia con autoanticuerpos positivos (principalmente anti-Ro/anti-La), lo que sugiere que los pacientes con SS inmunopositivos conforman un subgrupo con una mayor prevalencia de hemocitopenias.
La notable frecuencia de algunas manifestaciones hematológicas y su estrecha relación con manifestaciones clínicas e inmunológicas de SS subraya la importancia de dichas alteraciones como integrantes del amplio espectro de manifestaciones del SS primario, y su posible inclusión dentro de los criterios clasificatorios de SS debería considerarse en una futura revisión de los criterios actuales. Los resultados de la presente revisión sugieren determinar las distintas pruebas hematológicas de acuerdo con la sospecha diagnóstica. Un primer grupo de pruebas debería realizarse en pacientes con determinadas manifestaciones clínicas como infecciones repetidas (en los que solicitaremos una dosificación de Ig), o anemia aguda (en los que realizaremos el estudio de hemólisis o de anemia perniciosa). En cambio, podríamos considerar como pruebas hematológicas rutinarias (p. ej., realizadas anualmente) el hemograma, la VSG, el proteinograma y la inmunoelectroforesis sérica, debido a las frecuentes alteraciones halladas en dichas pruebas y a su estrecha relación con manifestaciones clínicas e inmunológicas características del SS (tabla 10).
