





High sensitivity and specificity of molecular biology techniques have proven usefulness for the detection, identification and typing of different pathogens. The ITS (Internal Transcribed Spacer) regions of the ribosomal DNA are highly conserved non-coding regions, and have been widely used in different studies including the determination of the genetic diversity of human fungal pathogens. This article wants to contribute to the understanding of the intra- and interspecific genetic diversity of isolates of the Histoplasma capsulatum and Sporothrix schenckii species complexes by an analysis of the available sequences of the ITS regions from different sequence databases. ITS1-5.8S-ITS2 sequences of each fungus, either deposited in GenBank, or from our research groups (registered in the Fungi Barcode of Life Database), were analyzed using the maximum likelihood (ML) method. ML analysis of the ITS sequences discriminated isolates from distant geographic origins and particular wild hosts, depending on the fungal species analyzed.
This manuscript is part of the series of works presented at the “V International Workshop: Molecular genetic approaches to the study of human pathogenic fungi” (Oaxaca, Mexico, 2012).
Las técnicas de biología molecular han proporcionado instrumentos de alta sensibilidad y especificidad, útiles para la detección, identificación y tipificación de diferentes patógenos. Las regiones ITS (Internal Transcribed Spacer) del ADN ribosómico están altamente conservadas y no son codificantes. Estas regiones se han utilizado ampliamente en diferentes tipos de estudios, incluida la determinación de la diversidad genética de hongos patógenos del ser humano. La finalidad de este artículo es contribuir al conocimiento de la diversidad genética intra- e interespecífica de aislamientos de los complejos de Histoplasma capsulatum y Sporothrix schenckii a través del análisis de las secuencias disponibles de las regiones ITS en distintos bancos de secuencias. Las secuencias de las regiones ITS1-5.8S-ITS2, de cada hongo, depositadas en el GenBank, junto con las obtenidas por nuestros grupos de investigación (depositadas en la Fungal Barcoding of Life Database), se analizaron con el método de máxima probabilidad (ML, por sus siglas en inglés). El análisis ML de las secuencias de las regiones ITS discriminó aislamientos de orígenes geográficos distantes y de huéspedes salvajes particulares, de acuerdo con la especie fúngica analizada.
Este artículo forma parte de una serie de estudios presentados en el «V International Workshop: Molecular genetic approaches to the study of human pathogenic fungi» (Oaxaca, México, 2012).
Reliable identification of pathogenic fungal species is fundamental to epidemiology in terms of biodiversity, geographical variation, and environmental changes. Species identification in fungi is particularly challenging because of their transient nature. Limitations to the studies of diversity in mammalian pathogenic fungi exist due to a lack of taxonomic specialists, and scarce and incomplete data for many taxonomic characters, which has been suggested by Suwannasai et al.17 and Tantichareon.19
Pheno- and genotyping of fungal strains have been used as important tools for identifying environmental sources of outbreaks as well as confirming the existence of pathogens in natural habitats. These different typing methods have used both conventional and molecular techniques.
Although phenotyping has continuously been used to study fungi, sensitive and specific genotyping methods are being developed to characterize fungal species, but different criteria must be met to be accepted by specialists. In many cases, genotyping methods compare DNA polymorphisms and classify fungal organisms according to the principles of molecular systematic. For Histoplasma capsulatum (etiological agent of the systemic mycosis histoplasmosis) and Sporothrix schenckii (etiological agent of the subcutaneous mycosis sporotrichosis) typing and classification, different molecular techniques have been applied, among them, various PCR methods using genomic sequences.7,8
There is a wide array of molecular markers for microorganism identification and genotyping or molecular classification. Among them, the Internal Transcribed Spacer (ITS) regions stand out for the study of closely related taxa, due to genetic diversity associated with the high rate of evolutionary changes characteristic of these regions.12 ITS consist of two variable non-coding regions (ITS1 and ITS2) inserted between the highly conserved small subunit 18S, the 5.8S, and the large subunit 28S of the rDNA gene cluster.12
ITS as a molecular target for fungal identification are supported by several unique characteristics: (i) The complete ITS region has a length between 600 and 800bp and can be easily amplified, using universal primers that are complementary to rDNA sequences. (ii) The multicopy nature of the repeat regions of the rDNA allows for the amplification of the ITS regions from small, diluted or degraded DNA samples. (iii) Several studies have demonstrated that the ITS regions are highly variable among morphologically distinct fungal species.12
The usefulness of ITS markers has been documented in several studies of phylogeny and genotyping of H. capsulatum1,5,6,11 and S. schenckii.2–4,22
The Mexican Barcode of Life project for the H. capsulatum and S. schenckii species complexesThe Mexican Barcode of Life (MEXBOL) resulted from the work of Mexican investigators as part of the international DNA barcoding (iBOL) project. MEXBOL is now part of a network with funding from the Consejo Nacional de Ciencia y Tecnología (CONACyT) and the Comisión Nacional para el Conocimiento y Uso de la Biodiversidad (CONABIO). The Natural Sciences and Engineering Research Council of Canada (NSERC) developed a Barcode of Life Database (BOLD) based on a specific informatics infrastructure. The cytochrome oxidase subunit 1 (COI), ribulose-bisphosphate carboxylase (rbcL), maturase K (matK), and ITS regions are among the Barcode sequences used. In addition to the assembly of barcode information and maintenance of these records by the BOLD system, a copy of all sequence and key specimen data is archived at the National Center for Biotechnology Information (NCBI) or its sister genomic repositories, the DNA Data Bank of Japan (DDBJ) and the European Molecular Biology Laboratory (EMBL), when results are ready for public release.13
The identification of H. capsulatum and S. schenckii isolates from different sources and origins by the sequences of the ITS regions started in 2010 as a project for the MEXBOL network for fungi. To date there are 19 ITS1-5.8S-ITS2 sequences of H. capsulatum from the Laboratorio de Inmunología de Hongos and 10 sequences of Sporothrix spp. from the Laboratorio de Micología Básica, Departamento de Microbiología y Parasitología, Facultad de Medicina, UNAM, deposited in the BOLD System. Sequences were obtained from isolates that were previously pheno- and genotypically well identified.10,14,15
Data regarding the natural hosts, sources, and samples of the 19 H. capsulatum and 10 Sporothrix spp. isolates are shown in Table 1. Fungal specimens are deposited in the Culture Collection of H. capsulatum from the Laboratorio de Inmunología de Hongos and the Culture Collection of Fungal Pathogens of the Laboratorio de Micología Básica, from the Departamento de Microbiología y Parasitología, Facultad de Medicina, UNAM. In addition, they are registered in the database of the World Federation for Culture Collection, with code number LIH-UNAM WDCM817 for H. capsulatum (http://www.wfcc.info/ccinfo/index.php/collection/by_id/817) and code number BMFM-UNAM WDCM834 for Sporothrix spp. (http://www.wfcc.info/ccinfo/index.php/collection/by_id/834).
Data of isolates of Histoplasma and Sporothrix complexes analyzed using the ITS1-5.8S-ITS2 region.
| Isolate | Host or source sample | Species | Barcode accession number |
| EH-53 | Human/Blood | H. capsulatum LAm A* | HIST001-13 |
| EH-315a | Bat/Intestine | H. capsulatum Lineage* | HIST002-13 |
| EH-317 | Human/Blood | H. capsulatum LAm A* | HIST003-13 |
| EH-373b | Bat/Lung | H. capsulatum LAm A* | HIST004-13 |
| EH-375b | Bat/Lung | H. capsulatum | HIST005-13 |
| EH-378b | Bat/Lung | H. capsulatum | **------ |
| EH-391c | Bat/Liver | H. capsulatum LAm A* | HIST006-13 |
| EH-393d | Bat/Spleen | H. capsulatum | HIST007-13 |
| EH-394Pd | Bat/Spleen | H. capsulatum | HIST008-13 |
| EH-398Pd | Bat/Lung | H. capsulatum | HIST009-13 |
| EH-449Ic | Bat/Intestine | H. capsulatum | HIST010-13 |
| EH-449Pc | Bat/Lung | H. capsulatum | HIST011-13 |
| EH-655Pe | Bat/Lung | H. capsulatum | HIST012-13 |
| EH-658He | Bat/Liver | H. capsulatum | HIST013-13 |
| EH-670Be | Bat/Spleen | H. capsulatum | HIST014-13 |
| EH-670He | Bat/Liver | H. capsulatum | HIST015-13 |
| EH-671Pe | Bat/Lung | H. capsulatum | HIST016-13 |
| EH-672Be | Bat/Spleen | H. capsulatum | HIST017-13 |
| EH-696Pe | Bat/Lung | H. capsulatum | HIST018-13 |
| EH-143 | Human/Cutaneous | S. schenckii | BMFM001-13 |
| EH-194 | Environmental/Rose plant | S. schenckii | BMFM002-13 |
| EH-195 | Environmental/Coffee soil | S. schenckii | BMFM003-13 |
| EH-197 | Human/Cutaneous | S. schenckii | BMFM004-13 |
| EH-230 | Human/Cutaneous | S. globosa | BMFM005-13 |
| EH-234 | Human/Cutaneous | S. schenckii | BMFM006-13 |
| EH-251 | Environmental/Soil | S. schenckii | BMFM007-13 |
| EH-252 | Environmental/Soil | S. schenckii | BMFM008-13 |
| EH-253 | Environmental/Soil | S. schenckii | BMFM009-13 |
| EH-254 | Environmental/Soil | S. schenckii | BMFM010-13 |
Current data from our laboratory teams, using evolutionary and genetic distance analyses by maximum likelihood (ML) of ITS1-5.8S-ITS2 sequences of H. capsulatum or Sporothrix spp. from the BOLD System and GenBank datasets, produced robust results to aid in understanding the similarities and diversities among isolates either of H. capsulatum or Sporothrix spp. from different sources and geographic origins.
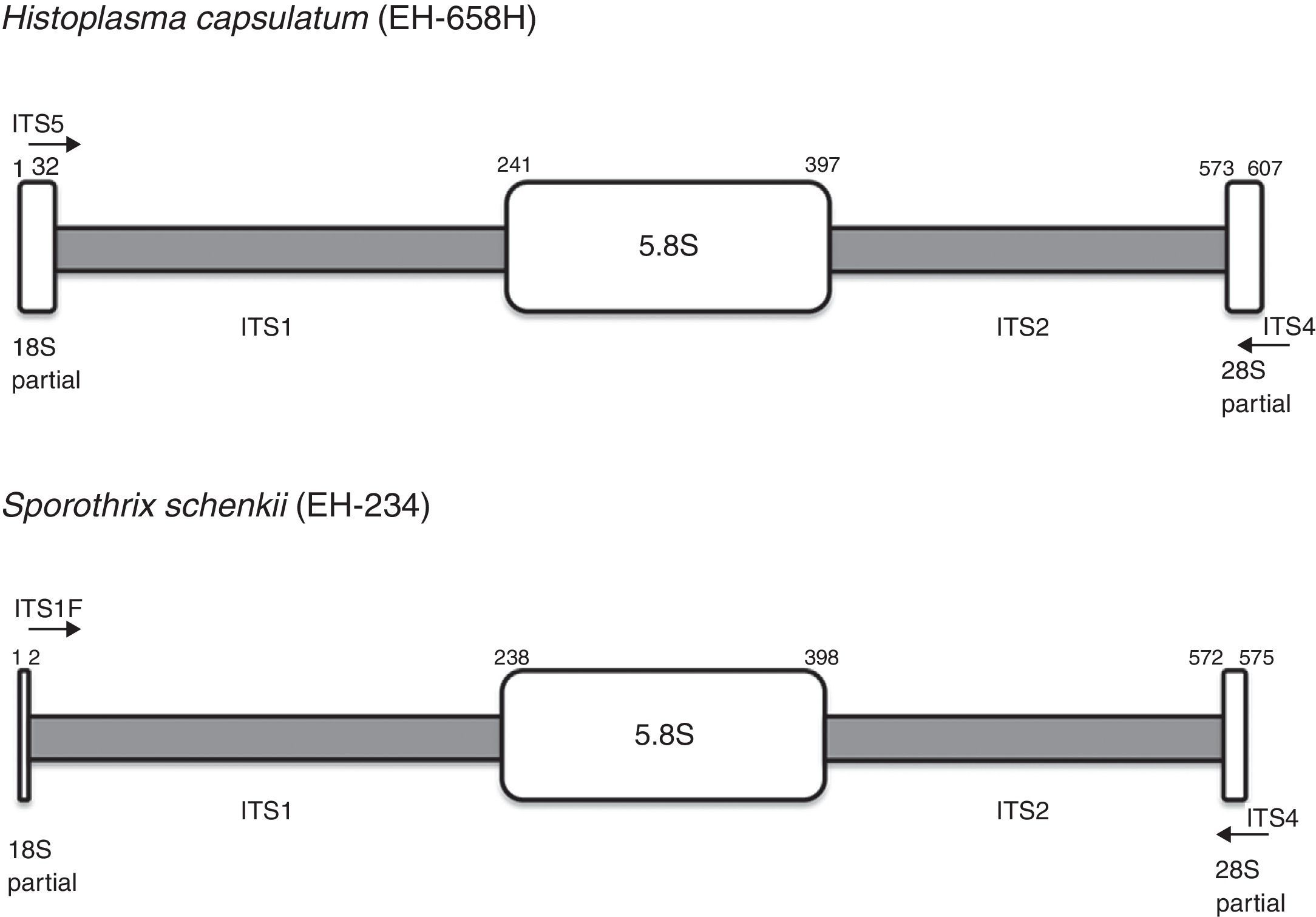
Sequences were generated by PCR assays with ITS5/ITS4 primers9 for H. capsulatum and ITS1F/ITS4 primers9 for Sporothrix spp. Fig. 1 shows the predicted products, 607bp for H. capsulatum and 575bp for Sporothrix spp, amplified by their respective primers. The ML trees generated are shown in Fig. 2.
 and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) for H. capsulatum, and ITS1F (CTTGGTCATTTAGAGGAAGTAA) and ITS4 for Sporothrix spp. The figure was modified from Saar and Polans,16 according to H. capsulatum and Sporothrix spp. ITS region data.")
ITS1-5.8S-ITS2 regions amplified from Histoplasma and Sporothrix samples. The schema depicts the regions amplified by the primers: ITS5 (5′-GGAAGTAAAAGTCGTAACAAGG-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) for H. capsulatum, and ITS1F (CTTGGTCATTTAGAGGAAGTAA) and ITS4 for Sporothrix spp. The figure was modified from Saar and Polans,16 according to H. capsulatum and Sporothrix spp. ITS region data.
 trees of ITS1-5.8S-ITS2 regions amplified for Histoplasma (A) and Sporothrix (B) samples. PCR products using primers aforementioned in Fig. 1 for each fungal species were sequenced and aligned by MUSCLE program (MEGA version 5). The best model of evolution to generate the ML phylogenetic trees was obtained by Tamura and Nei gamma distribution.18 A bootstrapping algorithm was achieved on the dataset for 1000 replicates, and values ≥70% were recorded for each tree node. BOLD System sequence accession numbers are referenced in table 1. GenBank sequences for both fungal species were included as reference strains or as outgroups in the ML analyses. Accession numbers are as follows: for H. capsulatum: H. capsulatum var. farciminosum (Hcf) H90 (AF322387.1), H. capsulatum var. capsulatum (Hcc) H81 (AF322385.1), H71 (AF322384.1), H62 (AF322379.1), H2 (AF322377.1), H9 (AF322378.1), H70 (AF322383.1), H68 (AF322382.1), H. capsulatum var. duboisii (Hcd) H88 (AF322386.1); for S. schenckii (Ss): ATCC 14284 (AF364061.1), ATCC 26331 (FJ545232.1), CNM-CM3477 (EU126945.1), CNM-CM3453 (EU126941.1), CNM-CM3462 (EU126943.1), CNM-CM3457 (EU126942.1), CNM-CM3450 (EU126940.1), Duke3751 (AB089138.1). The GenBank sequence of A. dermatitidis (Ad) ATCC 60915 (AF322388.1) was used as reference strain, and P. brasiliensis (Pb) IFM 41630 (AB304423.1) as the outgroup for H. capsulatum; O. fumeum (Of) CMW26818 (HM051415.1) was used as outgroup for Sporothrix spp. Parenthesis indicate H. capsulatum clades or lineages as Kasuga et al.7 Abbreviations: Sg=S. globosa; BE=Belgium; BR=Brazil; CO=Colombia; EG=Egypt; GT=Guatemala; JP=Japan; MX=Mexico; PA=Panama; USA=United States of America; ZM=Zambia.")
Maximum likelihood (ML) trees of ITS1-5.8S-ITS2 regions amplified for Histoplasma (A) and Sporothrix (B) samples. PCR products using primers aforementioned in Fig. 1 for each fungal species were sequenced and aligned by MUSCLE program (MEGA version 5). The best model of evolution to generate the ML phylogenetic trees was obtained by Tamura and Nei gamma distribution.18 A bootstrapping algorithm was achieved on the dataset for 1000 replicates, and values ≥70% were recorded for each tree node. BOLD System sequence accession numbers are referenced in table 1. GenBank sequences for both fungal species were included as reference strains or as outgroups in the ML analyses. Accession numbers are as follows: for H. capsulatum: H. capsulatum var. farciminosum (Hcf) H90 (AF322387.1), H. capsulatum var. capsulatum (Hcc) H81 (AF322385.1), H71 (AF322384.1), H62 (AF322379.1), H2 (AF322377.1), H9 (AF322378.1), H70 (AF322383.1), H68 (AF322382.1), H. capsulatum var. duboisii (Hcd) H88 (AF322386.1); for S. schenckii (Ss): ATCC 14284 (AF364061.1), ATCC 26331 (FJ545232.1), CNM-CM3477 (EU126945.1), CNM-CM3453 (EU126941.1), CNM-CM3462 (EU126943.1), CNM-CM3457 (EU126942.1), CNM-CM3450 (EU126940.1), Duke3751 (AB089138.1). The GenBank sequence of A. dermatitidis (Ad) ATCC 60915 (AF322388.1) was used as reference strain, and P. brasiliensis (Pb) IFM 41630 (AB304423.1) as the outgroup for H. capsulatum; O. fumeum (Of) CMW26818 (HM051415.1) was used as outgroup for Sporothrix spp. Parenthesis indicate H. capsulatum clades or lineages as Kasuga et al.7 Abbreviations: Sg=S. globosa; BE=Belgium; BR=Brazil; CO=Colombia; EG=Egypt; GT=Guatemala; JP=Japan; MX=Mexico; PA=Panama; USA=United States of America; ZM=Zambia.
Concerning H. capsulatum, Fig. 2A highlights the sequences of all isolates from different geographic origins and phylogenetic species that clustered together in a major group sustained by 99% of bootstrap values (BT). This finding confirms the high similarity of the isolates analyzed, separates a reference strain of Ajellomyces dermatitidis (nearby sister), and underlines the genetic distance from a heterologous pathogenic fungus, Paracoccidioides brasiliensis, used as an outgroup in the ML analysis. The ML tree topology of H. capsulatum sequences in Fig. 2A clearly confirms that inter-specific diversity among fungal pathogens that cause respiratory diseases is well sustained using ITS1-5.8S-ITS2 region sequence analyses. Besides, it should be emphasized that ITS could also distinguish intra-specific diversity among H. capsulatum isolates, evidenced by the formation of a subgroup sustained by 89% BT, which contains six fungal isolates recovered from a particular wild host (Tadarida brasiliensis bats), together with one isolate recovered from an infected Mormoops megalophylla bat. This conclusion is consistent with results for these particular isolates using other molecular markers.20,21 In contrast, the ITS1-5.8S-ITS2 region sequence analysis in Fig. 2A could not discriminate cryptic species or clades of the H. capsulatum complex (NAm 1, NAm 2, LAm A, LAm B, African, Eurasian, and some lineages) and the taxonomic varieties H. capsulatum var. farciminosum, H. capsulatum var. capsulatum, and H. capsulatum var. duboisii.
The tree generated for Sporothrix (Fig. 2B) shows three groups in relation with the outgroup, Ophiostoma fumeum. The first group was formed by two isolates from the United States of America, eight S. schenckii from Mexico, and one isolate of S. schenckii and one of Sporothrix globosa from Guatemala, with a BT of 93%. The second group was formed by five isolates of S. schenckii, all from Brazil, which was sustained by a BT of 100%. Finally, the third group included only one isolate of S. schenckii from Japan (Fig. 2B). Therefore, the ITS1-5.8S-ITS2 region sequence is a molecular marker that could discriminate Sporothrix species from different geographic regions; however, this marker could not discriminate between Sporothrix species.
ConclusionsITS1-5.8S-ITS2 region sequences deposited in different databases could be utilized as a broad molecular marker for inter- and intraspecific genetic diversity of the H. capsulatum and S. schenckii species complexes. The intraspecific diversity of this genetic region could discriminate H. capsulatum or Sporothrix isolates according to their geographic distribution and association with environmental sources. However, ITS regions were unable to distinguish neither H. capsulatum species nor Sporothrix spp. among their respective phylogenetic, biological and/or taxonomic species complexes.
Conflict of interestsThe authors have no conflict of interests.
MLT and CT thank the MEXBOL program of CONACyT-Mexico, ref. numbers: 122896 and 122481, respectively. RTA thanks MEXBOL program of CONACyT, ref. 122481, for his scholarship. The authors thank Ingrid Mascher for editorial assistance.
These authors participated in the design and coordination of the Mexican Thematic Net for Fungi Barcode of Life (MEXBOL) of the Consejo Nacional de Ciencia y Tecnología (CONACyT)-MEXICO and have equally contributed to this review.

